
INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a virus that first appeared in Wuhan at the end of 2019 and was the cause of the last major pandemic [1]. The world was paralyzed under conditions of biological protection that today serve as a lesson for future pandemics [2]. However, despite the confinements, the human losses due to this virus were innumerable [3].
SARS-CoV-2 is a highly transmissible coronavirus, similar to other highly pathogenic coronaviruses such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) [4]. Individuals who contract SARS-CoV-2 experience symptoms of viral pneumonia, including fever, cough, and chest pain. In more severe cases, they may present with difficulty breathing and bilateral lung infiltrates. Unlike SARS-CoV, it has been demonstrated that SARS-CoV-2 can be transmitted by individuals who may not exhibit symptoms [5].
SARS-CoV-2 belongs to the betacoronavirus 2B lineage and diverges from SARS-CoV. However, SARS-CoV-2 is a distinct virus with its own unique characteristics and genetic composition [6]. SARS-CoV-2 enters human cells by binding to angiotensin-converting enzyme 2 as its receptor.
The Mpro protein, an enzyme utilized by the virus to process essential proteins for replication, presents a potential target for inhibiting viral replication. Many researchers are actively seeking compounds capable of blocking Mpro activity, viewing it as a promising strategy in the treatment of COVID-19 [7,8].
Currently, several different types of vaccines have been developed, primarily based on messenger ribonucleic acid and viral vector technology [9]. However, despite successful vaccinations in several countries, significant breakthrough infections (infections after vaccination) occur due to various factors, including decreased antibody levels [10], the presence of new viral variants [11], and comorbidities such as cancer, immunodeficiencies, or conditions like diabetes, which have shown increased susceptibility to severe COVID-19 disease [12].
Currently, a Michael acceptor inhibitor known as N3 has been developed using computer-aided drug design. N3 exhibits specific inhibition of Mpro from multiple coronaviruses by targeting a highly conserved substrate recognition pocket shared by SARS-CoV, MERS-CoV, and infectious bronchitis viruses [7,13]. Additionally, substances like Sitagliptin and Melittin can inhibit Mpro in a manner similar to N3 [14].
Given this, it is critically important to develop effective and efficient medications that can be beneficial in managing moderate or severe disease. Worldwide efforts have been dedicated to inventing such drugs and identifying pre-existing medications capable of neutralizing the virus [15,16], as well as the search for antiviral compounds from medicinal plants [17]. Molnupiravir and Paxlovid (Nirmatrelvir/Ritonavir) have been developed and are utilized as emergency therapies for mild to moderate COVID-19. Molnupiravir is a ribonucleoside analog inhibitor of RNA-dependent RNA polymerase, nirmatrelvir covalently inhibits Mpro, ritonavir inhibits CYP3A to slow the metabolism of nirmatrelvir (and does not inhibit Mpro because human immunodeficiency virus and SARS-CoV-2 proteases are from different clans) [18–20], N3 also covalently inhibits Mpro [7,13], and sitagliptin does not inhibit Middle East respiratory syndrome (MERS) entry via Dipeptidyl peptidase 4 [21] and is a noncovalent inhibitor of Mpro. However, expanding the repertoire of small molecules specifically designed as chemotherapy agents against SARS-CoV-2 remains essential.
While it is true that covalent inhibitors are being developed for the treatment of COVID-19 [22], molecules that bind covalently may be more likely to trigger an immune response or cause toxicity due to the formation of covalent adducts with the nontarget proteins [23]. Furthermore, noncovalently linked molecules can be designed to have a wide range of pharmacokinetic and pharmacodynamic properties, which may be useful to optimize efficacy and minimize side effects [24].
Since Sitagliptin is a small molecule, our study utilizes it as a foundation to design new compounds using bioisosteric replacement techniques, aiming to create novel SARS-CoV-2 Mpro inhibitors that surpass the effectiveness of Sitagliptin.
METHODS
Bioisosteres design
The design of Sitagliptin derivatives was conducted using bioisosteric replacement techniques, aiming to discover novel compounds with enhanced activity and low toxicity. To achieve this were utilized the ZINC20 database [25] and SeeSAR program [26] to prepare the derivatives, modifying each part of sitagliptin (Fig. 1) based on the following parameters:
- The 1,2,4-Triazolo[4,3-a]pyrazine segment and the keto functionality of the 3-amino-butan-1-one chain were replaced, while the remainder of the molecule remained unchanged.
- The 1,2,4-Triazolo[4,3-a]pyrazine segment and the fluorine in the para position of 2,4,5-trifluorophenyl were preserved, while the remaining portion of the molecule was replaced.
- The 1,2,4-Triazolo[4,3-a]pyrazine segment and the fluorine in the meta position of 2,4,5-trifluorophenyl were preserved, while the remaining portion of the molecule was replaced.
- The trifluoromethyl group and the para fluorine of 2,4,5-trifluorophenyl were preserved, while the remaining portion of the molecule was replaced.
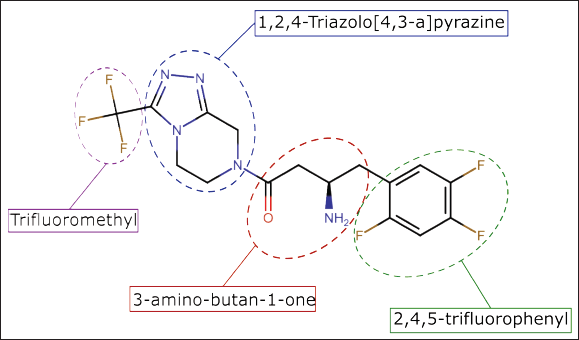 | Figure 1. Chemical structure and structural moties of sitagliptin. [Click here to view] |
The physicochemical properties of the resulting structures were computed using the SwissADME computer server [27]. By evaluating the relationship between molecular weight (MW), lipophilicity (LogP), and topological polar surface area (TPSA), compounds with the highest potential for gastrointestinal absorption and a reduced likelihood of crossing the blood-brain barrier were selected [28,29].
Protein target preparation
The SARS-CoV-2 protease Mpro was retrieved from the Protein Data Bank with the code 7BB2 [30]. The structure underwent a purification process, during which ions, ligands, and water molecules were eliminated using the PyMOL Molecular Graphics System 2.5 software program [31]. For molecular docking, the selected protein’s structure was parameterized using the AutoDock Tools program, polar hydrogens were added, nonpolar hydrogens were removed, and Kollman charges were assigned [32].
Molecular docking
Marvin v23.11, 2023, developed by ChemAxon (http://www.chemaxon.com), was used for drawing and characterizing chemical structures of bioisosteres and sitagliptin. Molecular docking was conducted using the Autodock Vina protocol [33], which employs the VINA genetic algorithm and predefined procedures for directed and rigid docking. The amino acid residues Glycine 143, Serine 144, Cysteine 145, Histidine 163, and Histidine 164 were identified as constituents of the potential binding site [34]. Subsequently, a grid box was created at the identified catalytic site with the following coordinates: center_x = -13.714, center_y = 17.195, center_z = 69.288, size_x = 18.0, size_y = 19.5, and size_z = 21.0. Each of the Sitagliptin derivatives was docked to this specific site on the S protein, revealing the most probable and energetically favorable binding conformations. The completeness was assessed 100-fold for each protein-ligand interaction.
The binding docking positions were visually examined for interactions using the software programs PyMOL [30] and Discovery Studio [34].
The most potent compounds were those with lower affinity energy and a greater number of hydrogen bond interactions and total interactions [35].
Pharmacokinetic properties
The pharmacokinetic properties of Sitagliptin derivatives were assessed using the pkCSM web server [36].
RESULTS
Based on the design parameters outlined in the Methods section, 10 bioisosteres of Sitagliptin were generated for each of the parameters A, B, and C. Additionally, 20 bioisosteres were obtained using parameter D, resulting in a total of 50 Sitagliptin-like molecules, as shown in Figure 2.
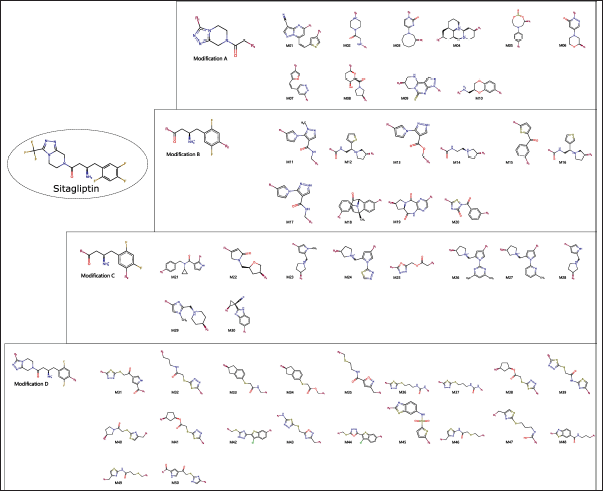 | Figure 2. Sitagliptin bioisosteres. [Click here to view] |
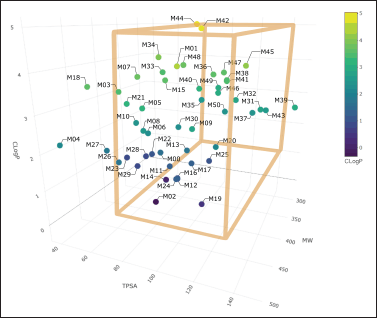 | Figure 3. Physicochemical properties of Sitagliptin bioisosteres. [Click here to view] |
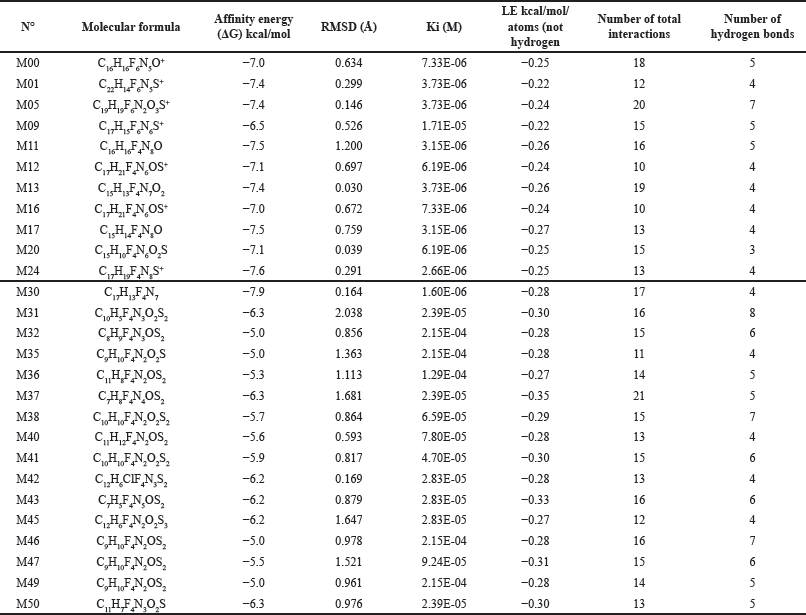 | Table 1. Affinity energy and number of interactions of Sitagliptin bioisosteres with anti-viral activity against SARS-CoV-2. [Click here to view] |
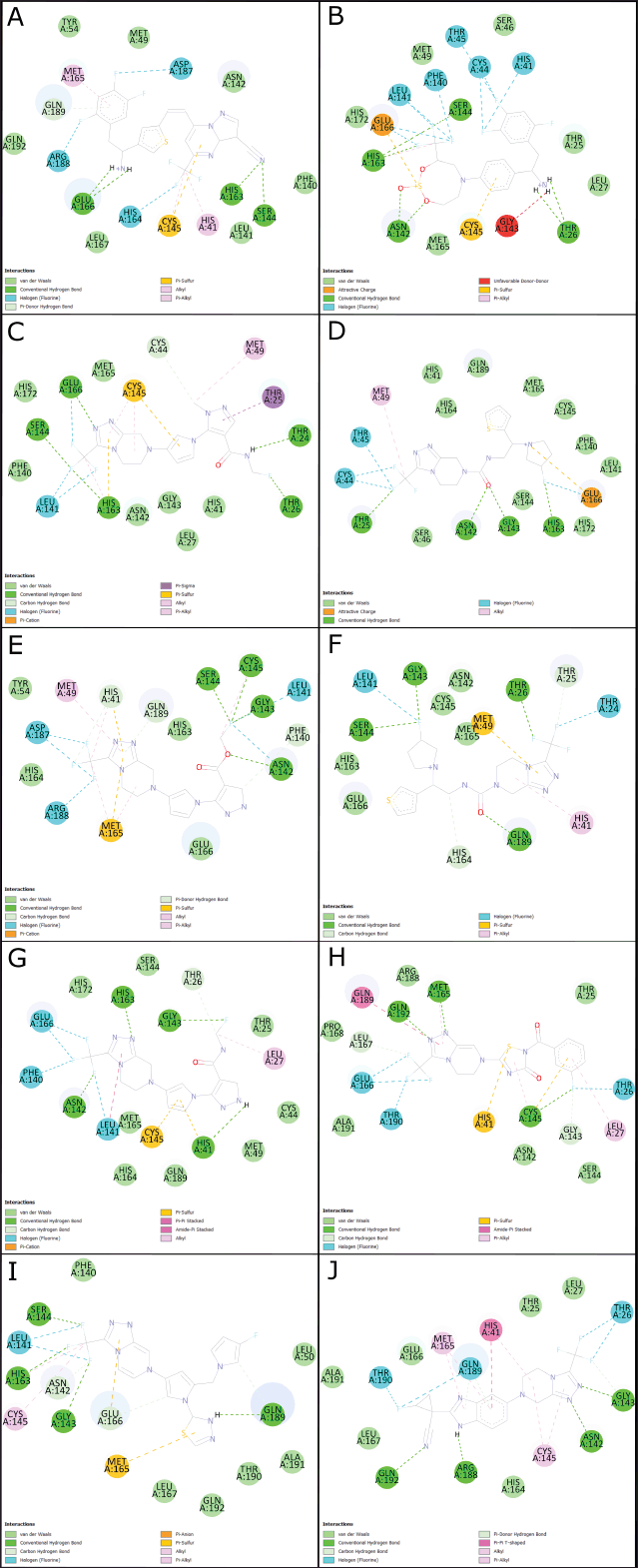 | Figure 4. 2D molecular docking of Sitagliptin bioisosteres with anti-viral activity against SARS-CoV-2. (A) Molecular docking of M01. (B) Molecular docking of M05. (C) Molecular docking of M11. (D) Molecular docking of M12. (E) Molecular docking of M13. (F) Molecular docking of M16. (G) Molecular docking of M17. (H) Molecular docking of M20. (I) Molecular docking of M24. (J) Molecular docking of M30. Source: BIOVIA Discovery Studio Program. [Click here to view] |
Similarly, the physicochemical properties of the 50 Sitagliptin bioisosteres were evaluated using the SwissADME web server. Key parameters, including MW, lipophilicity (CLogP), and TPSA, were three-dimensionally plotted in Figure 3. This analysis aimed to identify structures with improved gastrointestinal absorption and reduced likelihood of crossing the blood-brain barrier. Subsequently, the selected bioisosteres underwent molecular docking techniques for further assessment.
The molecular docking analysis was conducted on 26 bioisosteres of Sitagliptin that exhibited favorable characteristics related to gastrointestinal absorption and reduced brain distribution, which were maintained within a three-dimensional box. The results of the molecular docking (as shown in Table 1) were based on affinity energy (ΔG), ligand efficiency (LE), number of hydrogen bonds, and total interactions. Bioisosteres with lower energy and a greater number of interactions were considered suitable. Sitagliptin (M00) was also analyzed under the same conditions to serve as an in silico reference, with bioisosteres M01, M05, M11, M12, M13, M16, M17, M20, M24, and M30 exhibiting affinity energy lower than that of Sitagliptin. Their active site interactions are illustrated in Figure 4.
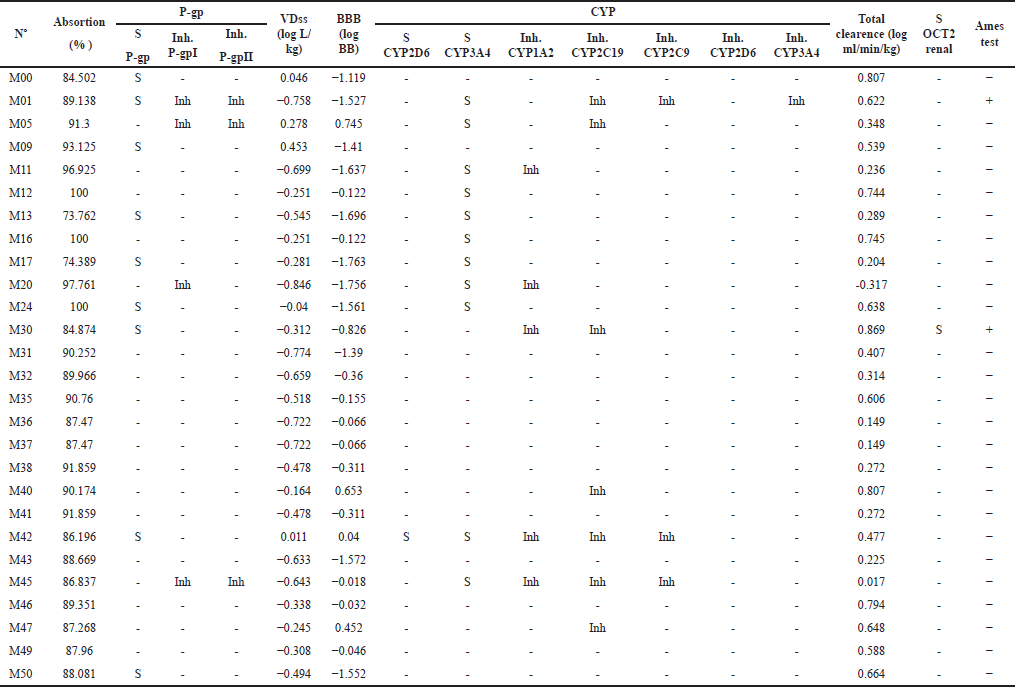
The pharmacokinetic properties of the 26 Sitagliptin bioisosteres were also assessed using the pkCSM web server, and the results are presented in Table 2. Among them, only the bioisosteres M01 and M30 tested positive for the Ames test, while M20 exhibited low renal elimination. Additionally, 17 bioisosteres did not inhibit any cytochrome P450 (CYP) or P-glycoprotein (P-gp).
 | Table 2. Pharmacokinetic properties of Sitagliptin bioisosteres with anti-viral activity against SARS-CoV-2. [Click here to view] |
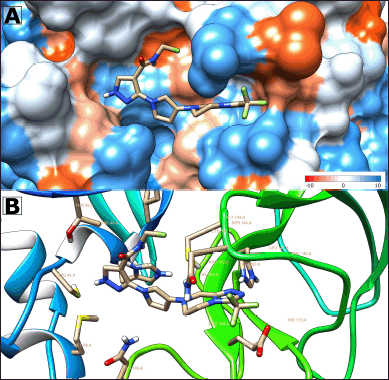 | Figure 5. 3D molecular docking of M17 bioisosteres with anti-viral activity against SARS-CoV-2. (A) Visualization of surface hydrophobicity of Mpro with M17 bioisister. (B) 3D representation of M17 with close residues of Mpro (less five angstroms). Source: UFC Chimera Program v1.16 (2021). [Click here to view] |
DISCUSSION
The ZINC20 database [25] and SeeSAR program [26] were used to generate 50 bioisosteres of sitagliptin. Among these, 26 molecules (in addition to sitagliptin) exhibit favorable gastrointestinal absorption and low distribution in the brain (as shown in Fig. 3). To assess oral bioavailability, key physicochemical characteristics were considered: CLogP, TPSA, and MW, following the guidelines set forth by Lipinski and Veber.
According to Lipinski [28], the CLogP values of these molecules should fall within the range of 1 to 5. This ensures that they are neither excessively lipophilic (which could reduce aqueous solubility) nor overly hydrophilic (which might hinder permeability). Additionally, the MW must be less than 500 Da to maintain the characteristics of small molecules.
Veber [29] further highlights the significance of TPSA. Molecules with a TPSA exceeding 140 Ų possess an excessive polar area, potentially forming numerous hydrogen bonds (as per Lipinski’s criteria). Consequently, their ability to traverse biological membranes decreases. Conversely, if the TPSA is less than 80 Ų, the molecules lack significant polar area and can readily enter the brain. However, for this study, drug candidates that primarily act at a peripheral level were sought.
Therefore, the molecules enclosed within the orange box in Figure 3 exhibit the most favorable physicochemical properties.
The molecular docking was performed using the 26 molecules exhibiting good oral absorption, in addition to Sitagliptin, which served as a reference. The concept of affinity energy refers to the force governing reversible interactions between two molecules (ligand and receptor). Lower energy requirements indicate a higher likelihood of successful interaction [37]. Although all the molecules displayed negative energy, implying potential binding to the active site, only 10 of them exhibited affinity energy less than −7.0—the same energy level observed for sitagliptin in this study. Consequently, these specific molecules may offer enhanced inhibitory activity against the Mpro of SARS-CoV-2.
Molecule M05 exhibits the highest number of hydrogen bonds, as depicted in Figure 4. However, it also displays unfavorable interactions that might constrain its effectiveness upon transitioning to in vitro or in vivo models. Additionally, M05 is the least rigid among the ten molecules, potentially leading to reduced selectivity [38]. Furthermore, its intricate and sizable ring structure could pose challenges to its synthetic viability [27].
The greatest number of highly active bioisosteres was obtained by retaining the 1,2,4-Triazolo[4,3-a]pyrazine moiety from the sitagliptin molecule. Therefore, new compounds should be tested while preserving this specific portion of the molecule. Among these compounds, M11, M17, M24, and M30 exhibit lower affinity energies compared to the remaining molecules.
Inhibition constant (ki) and LE are critical factors to consider. A lower affinity energy corresponds to a lower ki, signifying not only feasible interactions but also the favorable potency of these bioisosteres [37]. On the other hand, LE reflects the ligands’ ability to elicit a biological response upon binding to the target, quantified in terms of magnitude [39]. Mathematically, LE is calculated by dividing the affinity energy by the number of atoms (excluding hydrogen), thereby determining the energy force required for each atom to maintain interaction at the active site [40]. Table 1 shows that molecules M11, M13, M17, and M30 exhibit the lowest LE, with M17 and M30 having the most favorable values.
LE is considered favorable when values fall below −0.4. However, these thresholds vary across different drug discovery phases. For Hits, an LE of −0.41 is deemed suitable, while leading compounds in Phase II exhibit values between −0.39 and −0.42 [39]. Notably, molecules M17 and M30 possess LE values of −0.27 and −0.28, respectively. These values are suboptimal due to the ligands’ size. Binding efficiency correlates with molecular size—larger ligands tend to exhibit lower binding quality with the receptor [41]. Interestingly, despite deviating from this principle, the molecules resulting from our study demonstrate comparable or superior LE values compared to Sitagliptin. Moreover, these molecules hold promise for successive Structural Simplification studies, where structure size is minimized while maintaining or enhancing critical parameters. This approach allows for the formulation of substances with a higher likelihood of potency against the Mpro of SARS-CoV-2.
In addition to molecular docking, which enables us to infer pharmacodynamic properties in this study, pharmacokinetic properties were analyzed using the pkCSM server [36]. While binding energy and potency play crucial roles in the early stages of drug development, it is the pharmacokinetic properties and toxicity that ultimately determine the success of a studied molecule. Notably, a significant proportion of study compounds fail precisely due to unfavorable pharmacokinetic characteristics [42]. These properties aid in better selection, allowing us to identify molecules with the highest potential for future drug development. Table 2 presents the predicted pharmacokinetic properties of the 26 molecules (including Sitagliptin).
Among the molecules studied, namely M11, M13, M17, M24, and M30, none inhibit P-gp, which reduces the likelihood of interactions when co-administered with other drugs [43]. Conversely, their volume of distribution remains adequate, and they do not readily traverse the blood-brain barrier due to their logBB value being less than 0.3 [44], as predicted using the 3D box (Fig. 3). However, it is worth noting that molecules like M05 and M47 exhibit values above 0.3.
The inhibition of CYP enzymes is highly relevant in the context of drug–drug interactions. Therefore, caution must be exercised when inhibiting one or more CYP isoforms, particularly CYP2D6 and CYP3A4, which play a pivotal role in metabolizing a wide range of drugs [45]. Among the studied molecules, M11, M13, M17, M24, and M30 do not inhibit CYP2D6 or CYP3A4. However, M11 specifically inhibits CYP1A2, while M30 inhibits both CYP1A2 and CYP2C19. These findings warrant attention when considering the potential impact of these molecules.
Renal elimination is substantial for all molecules, with only M30 serving as a substrate for the organic cation transporter 2 (OCT2). OCT2 plays a crucial role in the initial step of renal secretion for organic cations: absorption from the blood, across the basolateral membrane, and into the renal proximal tubule cells. This process assumes significance when predicting potential drug–drug interactions [46]. While understanding the inhibition of this transporter is essential, knowledge of the substrates is equally relevant. Such understanding aids in comprehending the renal elimination process, as tubular secretion is modulated by the presence of OCT2.
The Ames mutagenicity test serves as a widely used method for toxicity assessment, employing bacteria to ascertain whether compounds exhibit potential carcinogenic properties [47]. Currently, computer algorithms aid in the in silico prediction of this assay [48]. Consequently, it has been established that molecules M1 and M30 are likely mutagenic substances.
Considering this, the M17 molecule (Fig. 5) exhibits both lower affinity energy and more favorable LE. Furthermore, its pharmacokinetic properties align well. Consequently, in vitro and in vivo studies could be conducted using this compound. Additionally, M17 serves as a promising foundation for designing new compounds, with careful consideration of the nuclei it contains.
CONCLUSION
In conclusion, M11 is a good candidate, but caution must be taken due to a probable inhibition of CYP1A2; M13, despite having affinity energy not as high as other molecules, has a higher LE, and its pharmacokinetic properties are adequate; M17 has better affinity energy and LE than M13, and has good pharmacokinetic properties. M24 has a higher affinity energy, but its LE is lower; additionally, its pharmacokinetic properties are adequate. Finally, M30, despite having the highest affinity energy and best LE, is probably a mutagenic molecule. Given this, the M17 molecule would be the best candidate to continue in in vitro or in vivo studies.
LIST OF ABBREVIATIONS
CYP: cytochrome P450; Inh: inhibition; ki: inhibition constant; LE: ligand efficiency; MERS: Middle East respiratory syndrome; MW: molecular weight; OCT2: organic cation transporter 2; P-gp: P-glycoprotein; S: substrate; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TPSA: topological polar surface area.
ACKNOWLEDGMENTS
The authors thank the Bioinformatics and Chemoinformatics Research Group (BIOQUIM) by the Universidad Nacional de Trujillo (Resolución Vicerrectoral de Investigación N°060-2023-VIN-UNT).
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Adil MT, Rahman R, Whitelaw D, Jain V, Al-Taan O, Rashid F, et al. SARS-CoV-2 and the pandemic of COVID-19. Postgrad Med J. 2021;97(1144):110–6. CrossRef
2. Khanna RC, Cicinelli MV, Gilbert SS, Honavar SG, Murthy GVS. COVID-19 pandemic: lessons learned and future directions. Indian J Ophthalmol. 2020;68(5):703. CrossRef
3. Sirleaf EJ, Clark H. Report of the independent panel for pandemic preparedness and response: making COVID-19 the last pandemic. Lancet. 2021;398(10295):101–3. CrossRef
4. Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2020;19(3):141–54. CrossRef
5. Johansson MA, Quandelacy TM, Kada S, Prasad PV, Steele M, Brooks JT, et al. SARS-CoV-2 transmission from people without COVID-19 symptoms. JAMA Netw Open. 2021;4(1):e2035057. CrossRef
6. Wang MY, Zhao R, Gao LJ, Gao XF, Wang DP, Cao JM. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol. 2020;10:587269. CrossRef
7. Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–93. CrossRef
8. Beltrán LA, De La Hoz-Rodríguez S, Iserte LB, Rodríguez S, Fernández-De-La-pradilla A, et al. Advances in the development of SARS-CoV-2 Mpro inhibitors. Molecules 2022;27(8):2523. CrossRef
9. Longo M, Scappaticcio L, Signoriello S, Caruso P, Maio A, Botta G, et al. Glucose control during breakthrough SARS-CoV-2 infections in vaccinated patients with type 1 diabetes. Diabetes Res Clin Pract. 2024;207:111044. CrossRef
10. Wheeler SE, Shurin GV, Yost M, Anderson A, Pinto L, Wells A, et al. Differential antibody response to mRNA COVID-19 vaccines in healthy subjects. Microbiol Spectr. 2021;9(1):10–1158. CrossRef
11. Abbasian MH, Mahmanzar M, Rahimian K, Mahdavi B, Tokhanbigli S, Moradi B, et al. Global landscape of SARS-CoV-2 mutations and conserved regions. J Transl Med. 2023;21(1):1–15. CrossRef
12. Röltgen K, Boyd SD. Antibody and B cell responses to SARS-CoV-2 infection and vaccination: the end of the beginning. Annu Rev Pathol. 2024;19(1):69–97. CrossRef
13. Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10):e324. CrossRef
14. Al-Rabia MW, Alhakamy NA, Ahmed OAA, Eljaaly K, Aloafi AL, Mostafa A, et al. Repurposing of sitagliptin- melittin optimized nanoformula against SARS-CoV-2; antiviral screening and molecular docking studies. Pharmaceutics. 2021;13(3):307. CrossRef
15. Talukder P, Saha A, Roy S, Ghosh G, Roy DD, Barua S. Drugs for COVID-19 treatment: a new challenge. Appl Biochem Biotechnol. 2023;195(6):3653–70. CrossRef
16. Gómez-Ríos D, López-Agudelo VA, Ramírez-Malule H. Repurposing antivirals as potential treatments for SARS-CoV-2: From SARS to COVID-19. J Appl Pharm Sci. 2020;10(5):001–9. CrossRef
17. Rasul HO, Thomas NV, Ghafour DD, Aziz BK, Salgado MG, Mendoza-Huizar LH et al. Searching possible SARS-CoV-2 main protease inhibitors in constituents from herbal medicines using in silico studies. J Biomol Struct Dyn. 2023;42:1–15. CrossRef
18. Hashemian SMR, Sheida A, Taghizadieh M, Memar MY, Hamblin MR, Bannazadeh Baghi H, et al. Paxlovid (Nirmatrelvir/Ritonavir): a new approach to Covid-19 therapy?. Biomedicine Pharmacother. 2023;162:114367. CrossRef
19. Strizki JM, Gaspar JM, Howe JA, Hutchins B, Mohri H, Nair MS, et al. Molnupiravir maintains antiviral activity against SARS-CoV-2 variants and exhibits a high barrier to the development of resistance. Antimicrob Agents Chemother. 2024;68(1):e0095323. CrossRef
20. Mahase E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;375:n2713. CrossRef
21. Raj VS, Mou H, Smits SL, Dekkers DHW, Müller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–4. CrossRef
22. Tang B, He F, Liu D, He F, Wu T, Fang M, et al. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. Biomolecules. 2022;12(6):746. CrossRef
23. De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur J Med Chem. 2017;138:96–114. CrossRef
24. Aljoundi A, Bjij I, El Rashedy A, Soliman MES. Covalent versus non-covalent enzyme inhibition: which route should we take? A justification of the good and bad from molecular modelling perspective. Protein J. 2020;39(2):97–105. CrossRef
25. Irwin JJ, Tang KG, Young J, Dandarchuluun C, Wong BR, Khurelbaatar M, et al. ZINC20-A free ultralarge-scale chemical database for ligand discovery. J Chem Inf Model. 2020;60(12):6065–73. CrossRef
26. SeeSAR version 13.0; BioSolveIT GmbH, Sankt Augustin, Germany; 2023 [cited 2023 Jun 19]. Available from: www.biosolveit.de/SeeSAR
27. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. CrossRef
28. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. CrossRef
29. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–23. CrossRef
30. Costanzi E, Kuzikov M, Esposito F, Albani S, Demitri N, Giabbai B, et al. Structural and biochemical analysis of the dual inhibition of mg-132 against sars-cov-2 main protease (Mpro/3clpro) and human cathepsin-l. Int J Mol Sci. 2021;22(21):11779. CrossRef
31. Schrödinger L, DeLano W. PyMOL [Internet]. 2020. Available from: http://www.pymol.org/pymol
32. Morris GM, Ruth H, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785. CrossRef
33. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. CrossRef
34. BIOVIA. Dassault systèmes BIOVIA discovery Studio v21.1.0.20298. San Diego, CA; 2020. Available from: https://www.3ds.com/products/biovia/reference-center
35. Rifaioglu AS, Atas H, Martin MJ, Cetin-Atalay R, Atalay V, Do?an T. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform. 2019;20(5):1878–912. CrossRef
36. Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–72. CrossRef
37. Michel J, Essex JW. Prediction of protein–ligand binding affinity by free energy simulations: assumptions, pitfalls and expectations. J Comput Aided Mol Des. 2010;24(8):639–58. CrossRef
38. Romera JA, Cortés-Cabrera Á, Sánchez-Murcia PA, Álvarez-Builla J, Gago F, María E, et al. Bases moleculares de la selectividad de ligandos por receptores de melatonina. Dianas. 2013;2(2):1–8. Available from: http://hdl.handle.net/10017/19004
39. Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–21. CrossRef
40. Horna-Rodriguez AM, Lopez-Gamboa JA, Silva-Correa CR, Antonio SGW, Gamarra-Sanchez CD, Villarreal-La Torre VE. In silico analysis of the polyphenolic metabolites of zea mays L. “Purple Corn” on HMG-CoA reductase. Pharmacogn J. 2022;14(3):549–58. CrossRef
41. Reynolds CH, Tounge BA, Bembenek SD. Ligand binding efficiency: trends, physical basis, and implications. J Med Chem. 2008;51(8):2432–8. CrossRef
42. Brito MA de. Pharmacokinetic study with computational tools in the medicinal chemistry course. Braz J Pharm Sci. 2011;47(4):797–805. CrossRef
43. Yu DK. The contribution of P-glycoprotein to pharmacokinetic drug-drug interactions. J Clin Pharmacol. 1999;39(12):1203–11. CrossRef
44. Clark DE. In silico prediction of blood–brain barrier permeation. Drug Discov Today. 2003;8(20):927–33. CrossRef
45. Hakkola J, Hukkanen J, Turpeinen M, Pelkonen O. Inhibition and induction of CYP enzymes in humans: an update. Arch Toxicol. 2020;94(11):3671–722. CrossRef
46. Belzer M, Morales M, Jagadish B, Mash EA, Wright SH. Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther. 2013;346(2):300–10. CrossRef
47. Zeiger E. The test that changed the world: the Ames test and the regulation of chemicals. Mutat Res Genet Toxicol Environ Mutagen. 2019;841:43–8. CrossRef
48. Modi S, Li J, Malcomber S, Moore C, Scott A, White A, et al. Integrated in silico approaches for the prediction of Ames test mutagenicity. J Comput Aided Mol Des. 2012;26(9):1017–33. CrossRef