
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease and is the primary cause of dementia [1], which can be described as a constant, and continuous loss of memory combined with cognitive impairment and change in personality. It is currently one of the chief causes of death and has affected more than 44 million people around the world [2]. AD mainly affects people of old age (>65 years), hence as the demographics of society changes, the prevalence of AD and other age-related dementias increases [3]. Discovery and development of drugs for AD is a tedious process [4]. In 2023 FDA had approved a new category of drug lecanemab, only for people with mild cognitive impairment however studies are stating the patients who received drug are experiencing edema, or fluid formation on the brain [5] Currently there are only six FDA (Food and Drug Administration) approved prescriptions drugs available to treat the symptoms associated with AD. These include Tacrine, Donepezil, Galantamine, and Rivastigmine which are acetylcholinesterase (AChE) inhibitors, Memantine which is an N–methyl-Daspartate receptor antagonist (NMDA), and Suvorexant which is an orexin receptor antagonist [6].
The brains of AD patients are characterized mainly by two histopathological features, which are amyloid (Aβ) plaques consisting of ß-amyloid (Aß) peptides located outside the cells and neurofibrillary tangles made up of hyper phosphorylated tau protein located within the neurons [7]. Evidence has emerged indicating that Aß peptide and its aggregation forms a vital aspect of the development of AD. BACE1 (ß-secretase) is an aspartic protease mainly present in the central nervous system (CNS) and contained in the presynaptic terminals [8]. It is responsible for generating Aß by breaking down the transmembrane protein APP (ß-amyloid precursor protein [9]. Usually, α-secretase governs proteolytic processing of APP in a healthy brain, however, when α-secretase is inhibited, ß- and γ-secretases take over and bring about the formation of neurotoxic Aß 1–40 and Aß 1–42 [10]. Aß 140 and Aß 1–42 aggregate easily and get deposited. These are the main components of Aβ plaques. The production and deposition of Aß are considered to be the factors leading to pathological changes in AD, such as neurofibrillary tangles, neuron loss, vascular injury, dementia, and many more [6].
BACE1 is considered a major target for the development of drugs that can decrease cerebral Aß levels for the treatment and prevention of AD. BACE1 inhibition would stop Aß production and curb the occurance of Aß-associated pathologies. Also, the studies on BACE1 knockout mice showed that the initial reports were free of any harsh phenotype and didn’t exhibit any pathological abnormalities [11]. Over the last few years, various BACE1 inhibitors underwent clinical assessment. Initially, many BACE1 inhibitors were discontinued during Phase 1 or early Phase 2 trials due to liver (LY2886721), ocular (LY2811376), or cardiac (AZD-3839) toxicity. Some BACE1 inhibitors such as verubecestat, atabecestat, lanabecestat, LY3202626, and umibecestat advanced well in clinical trials but failed to display improvement in cognition and function in placebo-oriented studies. Elenbecestat was the final BACE1 inhibitor to be discontinued in Phase 3 trials [8].
BACE1 is a transmembrane protein of the type 1 category. It consists of a sequence of 82 amino acids running C-terminally to the homologous pepsin carboxyl terminus, including a lumenal expansion, a hydrophobic site carrying the transmembrane and cytosolic areas. The lumenal extension spans up to about 35 amino acids and is comprised of secondary structures that directly interact with it by fixing to the pepsin globular catalytic region. Disulfide bonds are attached to both extremes of the lumenal expansion and are directly connected to the catalytic domain. A small sequence of about 11 amino acids secures the catalytic site of BACE1 to the lipid bilayer membrane, C-terminally of the last disul?de bonded cysteine of the luminal expansion. Distant from a hydrophobic region of length of 26 amino acids including the transmembrane area, another sequence of 21 amino acids expands into the cytoplasm (residues 494–501). This small stretch contains a dileucine chain that interacts with the cytosolic region of APP. BACE1 action can also be controlled by the location of the “beta ?ap”, a β-sheet hairpin loop that is present over the active site [12]. The active site of ß-secretase is located within the lumen of acidic intracellular compartments. BACE1 is resistant to the inhibitory effect of pepstatin and shows optimal activity at approximately pH 4.5 [11].
In this study, the aim was to carry out drug repurposing by identification of some existing small molecules of unrelated purpose, as potential BACE1 inhibitors using modern in silico tools of drug discovery. Here, we have considered natural compounds imported from ZINC database. Natural products refer to chemical compounds or substances produced by living organisms that is present in the nature. These can be classified into small molecules resulting from metabolic reactions and those obtained as a result of secondary or non-essential metabolism [13]. Natural products are associated with low toxicity [14]. In 2019, nearly 30% of drugs were approved in the US on the basis of drug repurposing. This practice of drug repurposing has previously been adopted for various disorders such as cardiovascular problems, cancer, obesity, erectile dysfunction, ceasing smoking habits, stress, psychosis, and many other health problems. Repurposing of drugs decreases the time and cost required for initial screening, toxicity analysis, clinical trials, large-scale manufacturing, and formulation process [15]. In silico-based studies provide much acceptable and results. Although in-silicon drug repurposing methods also have their own advantages, on the other side it has few disadvantages and limitations also. Limitations are related to the data we select for the study. If the volume of data is very less, a proper model cannot be generated, second, it is difficult to understand vague descriptions, third a lack of comprehensive data. These challenges are associated with drug repurposing methods but can be controlled by software engineering techniques.
MATERIALS AND METHODS
The study was conducted using Schrodinger’s Maestro Molecular platform (Version 12.1) (Schrodinger, LLC, New York) molecular docking and stimulation studies on the co-crystallized protein 4-LXM were carried out on a HP desktop with Linux (Ubuntu, version 18.04.1) as the operating system. The following tools like Protein Preparation Wizard, LigPrep, Glide, induced fit docking, MM-GBSA, QikProp, Desmond was used in this study.
Protein preparation
From the Research Collaboratory for Structural Bioinformatics Protein Data Bank, the protein structure PBD ID 4LXM was downloaded. This is an X-Ray diffraction structure of human Beta secretase which has a compound (1S,3S,4S,5R)-3-{4-amino-3-fluoro-5-[(1,1,1,3,3,3-hexafluoropropan-2-yl)oxy]benzyl}-5-[(3-tert-butylbenzyl)amino]tetrahydro-2H-thiopyran-4-ol 1-oxide(CHEMBL2425609) complexed to it. The co-crystallized protein structure had a resolution of 2.30 Å. Once the protein was downloaded then using the protein preparation wizard of the Schrödinger suit, the protein preparation process was carried out. In this preparation, the downloaded protein was refined and then reviewed and modified by removing all solvents (mostly water) except the ones that were found to be interacting with the active site and within 5 Å were retained and hydrogens molecules were added. Low-energy protein molecules are stable, therefore using OPLS3e (Optimized potential for liquid stimulation) energy minimized protein molecule was generated by the energy minimization process of the wizard. Missing chains in the downloaded protein structure were incorporated using the prime module of the protein preparation wizard [15,16].
Receptor grid generation
The co-crystallized ligand (1S,3S,4S,5R)-3-{4-amino-3-fluoro-5-[(1,1,1,3,3,3-hexafluoropropan-2-yl)oxy] benzyl}-5-[(3-tert-butylbenzyl)amino]tetrahydro-2H-thiopyran-4-o1-oxide binding site was identified as the active site. The active site of the co-crystallized ligand was taken as the centroid and by using the Grid-based ligand docking with energetics (GLIDE) module receptor grid box was generated [15,16]. The size of the grid box was of 10 × 10 × 10? [radius] and the distance between the grid points was set to a default of 2Å [16,17].
Ligand preparation
The compounds from the zinc database were downloaded. These structures were subjected to pre-processing using the LigPrep module of Schrodinger. This led to the generation of 3-D structures at pH 7.0 ± 2.0 under the OPLS3e force field [15,16]. This step basically adds hydrogens, converts structures from 2-D to 3-D, makes the bond lengths and angles realistic, and reduces the energy of the structures with correct parameters such as their ring conformations, chiralities, tautomers, stereochemistry, and ionization states [18].
Ligand docking
The docking of the Lig-Prep zinc database compounds to the receptor was carried out using the GLIDE tool [15,16]. The GLIDE tool determines the binding capability of the compound to the protein and finds the favorable interactions between the protein and the ligand, using this information it also assigns the docking score for each compound. The GLIDE tool also organizes the compounds in the decreasing order of their docking score. The tool has different scoring functions based on their precision namely High-Throughput Virtual Screening (HTVS), Standard precision (SP), and extra precision (XP) [most accurate]. First, all the drugs were screened via HTVS, and the best molecules were selected for further analysis by SP and XP screening, respectively, as HTVS is less accurate [15,16,19].
Binding energy calculation
The GLIDE docking tool helps us to determine if the protein interacts with the ligand but the timeperiod for which the interaction lasts [absolute binding] is dependent on the binding energy which is determined by using the prime module [20]. The prime module uses the Molecular Mechanics energies generalized Born and surface area solvation (MM/GBSA) method. This method depends on the Variable-dielectric generalized Bron salvation model which uses water as a solvent under the OPLS3e field and thereby calculates the binding energy of the interactions [15,16]. The top eight compounds according to their docking score after performing XP docking were selected and these compounds were subjected to MM/GBSA.
ADMET analysis
The analysis of absorption distribution metabolism elimination (ADME) was performed using the QikProp tool of the maestro model. ADME is a measure of the drug likeness of a compound. Thus, prediction of ADME properties through in silico approach has emerged as an attractive and cost-saving method. The Lipinski rule was followed for assessing the drug likeness of molecules. It is expected that it helps in reducing failure rate in drug development process and drug recalling. Also, the gap is decreased between R&D to market and cost is reduced at later stage of drug development. Although this is a well-established method for filtering suitable molecules but on the other hand it is unable to predict whether the compound would be effective as a therapeutic agent or not. The QikProp tool was used to predict various properties such as molecular weight of the compounds (MW), octanol/ water partition coefficient (QPlogPo/w), the number of hydrogen bond donors (donorHB), hydrogen bond acceptors (acceptHB), predicted brain/blood partition coefficient (QPlogBB), Polar Surface Area (PSA), Lipinski’s rule of 5 [15,16]. Default settings were employed for these calculations. The compound’s bioavailability relies on its absorption process and first-pass liver metabolism. The absorption of the compounds, on the other hand, is determined by their solubility and permeability characteristics and also how the compound interacts with the transporting system and enzymes responsible for the metabolism in the gut wall. The parameter to assess oral absorption of the molecules is the QPlogS i.e., Predicted aqueous solubility. The toxicity studies of top-hit molecules were executed through ADMETlab2.0 integrated online platform.
Induced fit docking
Based on the docking score (XP), MMGBSA calculations, and the druggability analysis, eight compounds were selected for IFD (Induced Fit Docking) XP, the process uses Glide and Prime modules which accurately predicts the behavior of the ligand inside a receptor which includes the ligands modes of binding and its structural movements in the receptor. The ligand undergoes conformational changes when it binds to the protein pocket which allows the ligand to bind better. The prepared protein was included in the workspace and then ligands were selected in the project table. Standard protocol was followed which generates up to 20 poses and van der Waal’s scaling was done at a default factor of 0.50 [16,18]. Finally, IFD score was generated, this score is generated as a function of the docking score, glide energy, glide emodel values, and types of interactions [21].
Molecular dynamics (MD)
MD studies were carried out using Desmond module from Schrodinger on a workstation having specifications of the desktop PC; Operating system (Linux), Version (Ubuntu 18.04.5 LTS), Graphics card (Intel HD graphics 2500 (IVB GT1), Processor (Intel core i3-3240 processor), RAM of 8GB [20,21]. Compound ZINC000150351431 (Phytosulfokine B) along with ZINC000069488328, ZINC000014945921 dock complex was selected on the basis of the previous results and was further subjected for MD at 100ns. The co crystallized ligand was also subjected to the MD simulation studies as a reference compound. MD included following steps; initially the complex of protein and the docked ligand were selected, then a periodic boundary condition of size 10 × 10 × 10 Å with an orthorhombic shape was created around it and the system was made isoosmotic by adding Na ions using System Builder [22]. The solvent model selected wad simple point charge. The system m?del w?s subje?ted t? energy minimiz?ti?n until ? gr?dient thresh?ld re??hed 25 k??l/m?l/? ?nd the system w?s b?l?n?ed ?t ? tem?er?ture ?f 300K ?nd 1 b?r ?ressure vi? N?T (Normal temperature and Pressure) ensemble [15,16]. The solvated system was minimized using the maximum iterations of 2,000 and 1.0 (kcal/mol/Å) convergence threshold for 100 ns. MD simul?ti?n w?s ?erf?rmed ?t 300K ?t ?tm?s?heri? ?ressure ?f 1.01325B?r ?nd ? t?t?l ?f 1,000 fr?mes were re??rded during the 100-nse? simul?ti?n. Simul?ti?n inter??ti?n di?gr?m w?s used for further analysis of MD results [23].
RESULTS AND DISCUSSION
Based on molecular docking, ADME, and MMGBSA, eight hit compounds were selected from a virtual screening of 35,711 natural compounds. The binding affinity (docking-score) of these compounds was higher than that of the co crystallized ligand. The interactions of co-crystallized ligand with the protein and the top compounds interactions were compared at the particular binding site.
Molecular docking
In the present study, ZINC database was used to download the dataset. Here, three lakh molecules were screened after applying Log P filter. The docking process was done by using different modes available in the GLIDE panel. The selected compounds from the Zinc database chosen were subjected for docking in HTVS mode, which summed up to 35,711 compounds. HTVS is used when a large number of compounds are to be screened as it is less time taking compared to SP and XP docking. Conformational sampling in HTVS mode is much more constrained than SP docking, and it cannot be used with score-in-place. The top 10 % of the resultant compounds from HTVS docking were filtered based on their dock score and were further subjected for docking in SP mode, which totaled up to 4,117 compounds. SP docking maintains a balance between speed and accuracy. The top 10 % of the resultant compounds from SP docking were further subjected for docking in XP mode, which totaled up to 509 compounds. XP docking uses descriptors and explicit water technology, is most accurate but time taking and is used to elude any chances of false-positive results and therefore the resultant compounds from this docking ensure a proper correlation between the docking score and binding pose of drugs [24]. Finally, the top eight compounds were chosen based on their XP docking score and protein–ligand interactions. All of the eight compounds had a docking score ranging between –12.333 Kcal/mol and –11.206 Kcal/mol. BACE1’s binding pocket is made up of three main components: a, the catalytic aspartic acid residues that are important for BACE1’s proteolytic activity; B, the flap, which is the most flexible part of the binding site and regulates substrate access; and C, the 10 s loop near the S3 pocket. A: The catalytic dyad of Asp32 and Asp228 is found in the ligand binding sites and is important for the enzyme’s proteolytic action. As a ligand binds to these two amino acids, it increases its binding affinity and, as a result, its potency [25]. H-bond with GLN73, in addition to the aspartic dyad, was discovered to be essential for inhibition [10]. All the interactions like H-bond, hydrophobic interactions, polar interactions, π-cation, and charged positive and negative interactions for nine hits have been summarized in Table 1.
Out of the top ligands and the cocrystallizedstructure, ZINC000069488328, ZINC000049089131, ZINC000261496860 made Hydrogenbonding interactions with the Aspartic Dyads ASP228 and ASP32, as well as GLN73, while ZINC000150351431, ZINC000014945921, ZINC000069488195, CHEMBL2425609(cocrystallised ligand), made H-bond interactions with only the aspartic dyad except for ZINC000040164523, which formed H-bonding with ASP228 but not with ASP32. Other common hydrophobic residues were found to be ILE118, TYR71, TYR198, and LEU30. The cocrystal exhibited aromatic π-π stacking interactions with TYR71, with only two out of the eight ligands, specifically ZINC000150351431 and ZINC000049089131, displaying this characteristic. However, aromatic π-π stacking involving ZINC000069488328 was observed in conjunction with PHE108. Cocrystallised ligand (CHEMBL2425609) is exhibiting hydrophobic interactions with amino acids such as TYR115, ILE118, LEU30, ILE126, ILE226, TYR198, VAL69, PRO70, TYR71, VAL332, ILE110, and PHE108. CHEMBL2425609 and ligands displayed similar hydrophobic interactions with specific amino acids, including ILE118, TYR71, TYR198, and LEU30.
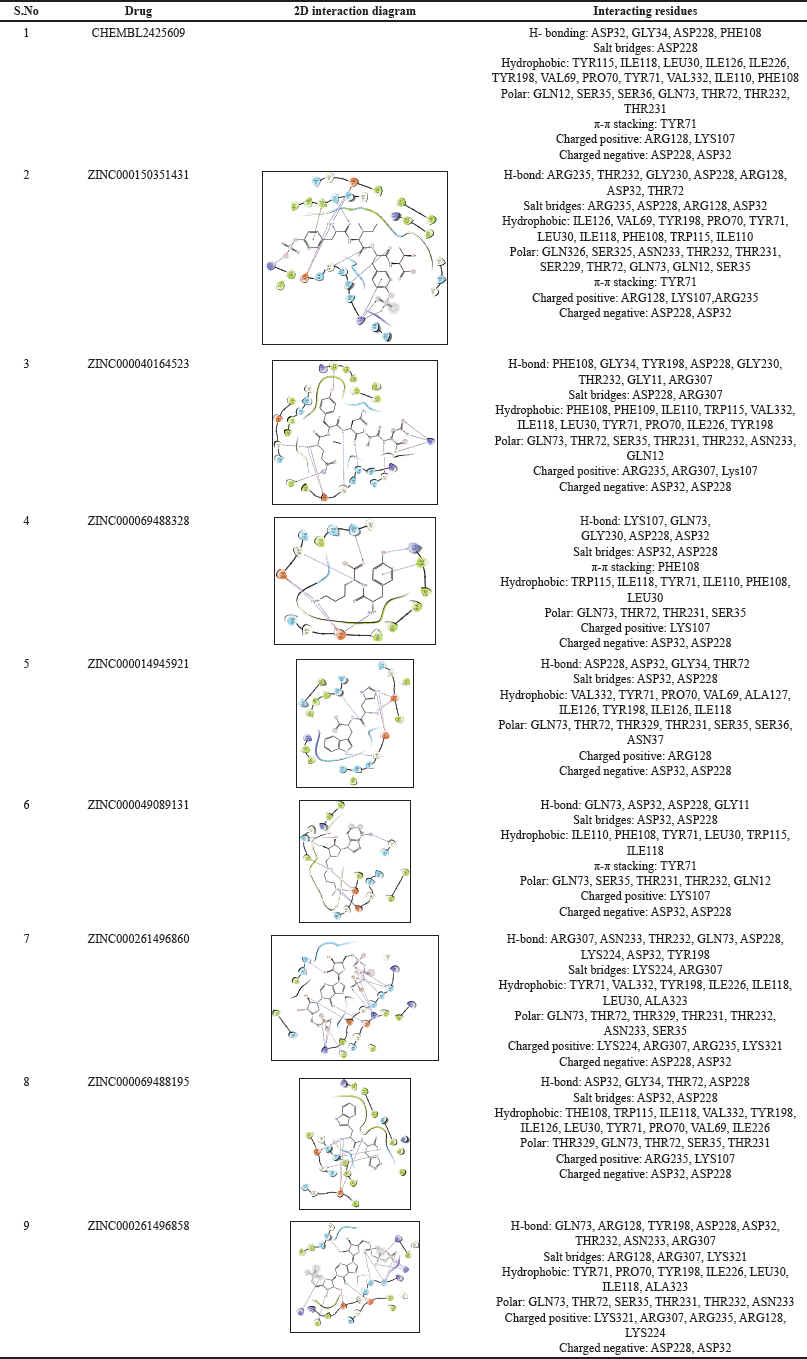 | Table 1. 2-D interaction diagrams with an illustration of all interacting residues. [Click here to view] |
MMGBSA
The selected compounds were subjected to prime MMGBSA analysis in selected docked poses to measure the protein-ligand complex stability in the form of binding energy and their dG binding energy was found to be > –47 kcal/mol (Table 2). The dG binding energy of the cocrystallised drug CHEMBL2425609 was discovered to be –103.74 kcal/mol. Therefore, these results suggest that all the ligands show stability in their docked poses and can act as potential BACE 1 inhibitors. ZINC000150351431 showed the most favorable binding energy of –106.63 kcal/mol.
ADMET analysis
ADME properties of the top nine hits were studied using the QikProp module. The ADME properties included molecular weight, total solvent accessible surface area, number of H-bond donors, number of H-bond acceptor, predicted octanol/water partition coefficient (Log Po/w), predicted aqueous solubility (QPLog S), predicted blood/brain partition coefficient (Log BB), PSA and Lipinski rule of five (Table 3). The rule of five stipulates that no more than one of the five properties of an orally active medication can be violated [26]. Only four hits were in accordance with this rule ZINC000069488328, ZINC000014945921, ZINC000049089131, and ZINC000261496858. The co-crystallised ligand was found to violate the rule of five.
One of the most important elements in drug development for CNS illnesses is the presence of a blood–brain barrier (BBB) that limits the movement of chemicals to the brain. A compound must have significant properties like QlogBB and PSA to be regarded as a CNS leading candidate. For a drug to pass the BBB, which is required for Alzheimer’s treatment, the QlogBB value must fall within the agreed-upon range of –3.0–1.2. Many researchers have employed PSA as a predictor of BBB penetration [26]. In addition, the prediction of binding to human serum albumin was also taken into account. After thorough analysis, it was found that only ZINC000069488328 was in accordance to the range of QPlogKhsa for prediction of binding to human serum albumin, QlogBB for BBB penetration, and PSA. QP log S values for most of the compounds were acceptable. Oral bioavailability (F ) results (Table 4) were found to be favorable for ZINC000069488328, ZINC000014945921, ZINC000150351431, ZINC000694881954 and co-crystallized ligand. The remaining compounds showed very poor bioavailability. AMES test (Salmonella typhimurium reverse mutation assay) results indicate that none of the top hit molecules were toxic, also 0 alerts were found for acute toxicity results of these molecules.
Induced fit docking
Traditionally, ligands are docked into the binding site of the receptor while the receptor is kept stiff and the ligand is free to move in virtual docking experiments. Because proteins in the body naturally undergo side-chain or backbone movement, or both, when they connect to a ligand, therefore presuming a rigid receptor can lead to erroneous conclusions. These changes allow the receptor to better adjust its binding site to the shape and binding mode of the ligand. A glide application known as induced fit docking may be used to reliably estimate the binding affinity of new inhibitors to the produced protein crystal, simulating this process. It is primarily used to develop an accurate complex structure for a ligand that is known to be active but cannot dock in a receptor’s rigid structure, and false negatives can be filtered out by using additional conformations obtained with the induced fit protocol rather than screening against a single receptor conformation. It promotes the docking of ligands in various conformations and determines the better binding capacity of the ligand [27]. During this study, the ligand and van der Waals scaling was kept constant at 0.50. The calculations were performed by following the SP protocol. A maximum of 20 poses of the protein with ligands were generated and each pose was analyzed thoroughly. The pose that showed maximum interactions with required residues was further taken for MD studies. ZINC000040164523 showed the highest IFD score (–807.13) amongst the top nine compounds (Table 5). 3-D interactions of the few compounds are shown in (Fig. 1)
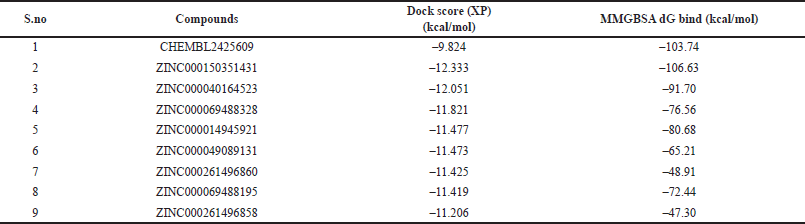 | Table 2. Prime MMGBSA score and docking score of top nine compounds. [Click here to view] |
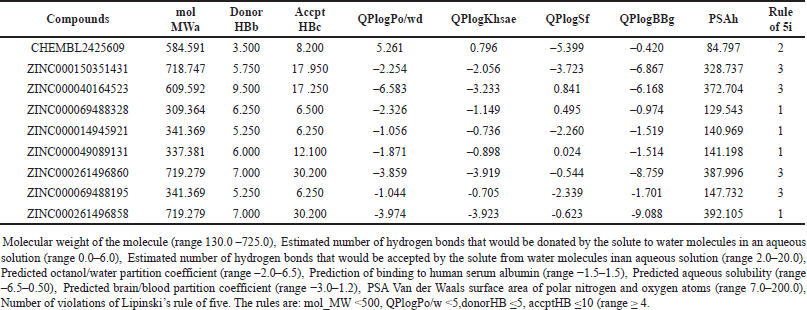 | Table 3. Analysis of ADME of the top nine compounds using QikProp. [Click here to view] |
 | Table 4. Toxicity prediction. [Click here to view] |
MOLECULAR DYNAMICS
MD is utilized to model ligand-protein complexes when physiologically relevant structures are present. MD stimulation has the advantage of accurately reflecting the real-world conditions of the biological system. Throughout the protein, explicit solvent representation is visible. It forms a highly dynamic protein structure, and the ligand protein complex is solvated with water in the same way that it is in nature. To obtain insight into binding stability and interactions with key amino acids within BACE 1 protein’s drug-binding pocket in a dynamic state. MD simulations were performed for three ligand-protein complexes viz., ZINC000150351431-BACE 1 docked complex (complex 1), ZINC000014945921-BACE 1 docked complex (complex 2), ZINC000069488328-BACE 1 docked complex (complex 3) based on molecular docking score, binding energy, IFD score, and oral bioavailability studies. ZINC000049089131, ZINC000261496860, ZINC000069488195, and ZINC000261496858 showed comparatively less binding energy to the target protein. ZINC000040164523 was found to have poor oral bioavailability. This study was also conducted for the co-crystallized ligand to compare the stability of the ligand-protein complex with the CHEMBL2425609 – protein complex. The frame was captured for 100ps in MD simulation, resulting in 1,000 frames being produced for a 100ns stimulation period and saved in a trajectory. In addition, the stability of the ligand-protein complex was estimated using an RMSD plot (Root mean square deviation) for BACE 1 (4LXM) protein and ‘Lig Fit Prot’ for the ligands.
For Complex 1 the protein and ligand RMSD values were determined to be within the range of 1.17 Å – 2.00 Å and 1.02 – 4.26, respectively. The complex was stable throughout the study, but a slight drift was observed for a period of 79.6 – 82 ns after which the complex stabilized in the later part of the study (Fig. 2 a). For complex 2, the protein and ligand RMSD values were determined to be within the range of 1.04 Å – 2.07 Å and 1.15 Å – 8.41 Å, respectively, major drifts were observed for 5 – 43 ns and 60 – 100 ns, and during this period the RMSD values were found to be more than the acceptable range of 1–3 Å (Fig. 2b). For complex 3 the protein and ligand RMSD values were determined to be within the range of 1.02 Å – 1.77 Å and 1.18 Å – 5.81 Å, respectively, initial drift from 0–11 ns was observed due to the initial stabilization of the protein structure with respect to ZINC000069488328 – protein complex and a major drift was observed from 51–53 ns after which the complex stabilized towards the end of the study (Fig. 2c). For co-crystallised ligand – BACE 1 complex, the protein and ligand RMSD values were determined to be within the range of 1.14 Å – 2.02 Å and 1.14 Å – 2.02 Å, respectively, and slight drift was observed at 93 ns after which the complex stabilized (Fig. 2d).
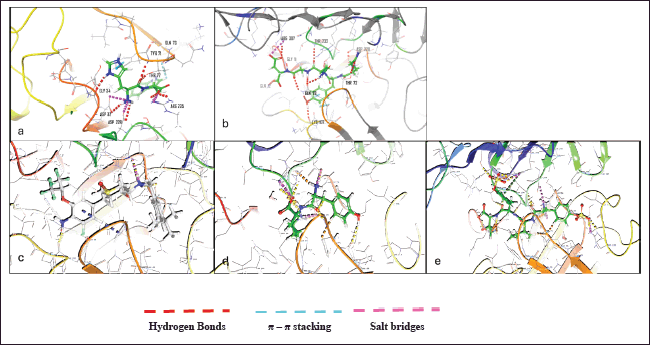 | Figure 1. 3-D Induced fit docking ligand interactions of a) ZINC000040164523 b) ZINC000014945921 c) CHEMBL2425609 d) ZINC000069488328 e) ZINC000150351431 BACE 1 (4LXM) protein and ‘Lig Fit Prot’ for the ligands. [Click here to view] |
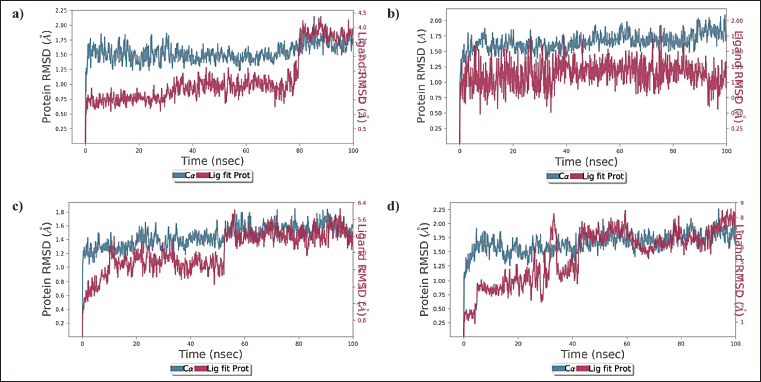 | Figure 2. a) RMSD plot of ZINC000150351431 and BACE 1 protein complex b) RMSD plot of ZINC000014945921 and BACE 1 protein complex c) RMSD plot of ZINC000069488328 and BACE 1 protein complex d) RMSD plot of CHEMBL2425609 (co-crystallised ligand) and BACE 1 protein complex. [Click here to view] |
Throughout the simulation study, the protein-ligand interactions were also monitored, and an analysis report was generated for the potential protein-ligand interactions. In the chosen trajectory, interactions that occur more than 30.0% of the simulation time were noted. Complex 1 made hydrogen bond interactions with ARG128, SER325, ARG235, GLN73, THR232, GLY230, THR231, ASP228, ASP32, and THR72. The π-cation interaction was made with ARG235. Hydrophobic interactions were also present but with weaker occupancy with LEU30, TYR71, and ARG235 (Fig. 3a). Complex 2 made hydrogen bond interactions with Thr232 and Asp32. Water bridged interactions were seen with LYS107 and THR72, also hydrophobic interactions with weaker occupancy were seen with LEU30, TYR71, PHE108, ILE110, and TRP115 (Fig. 3b). Complex 3 made hydrogen bond interactions with Thr72, Asp32, ASP228, and PRO70. It made water bridges interaction with ASP228 and π-π stacking interactions with TYR71. Hydrophobic interactions were observed with TYR71 and PRO70 (Fig. 3c). Co-crystallised ligand–BACE 1 complex made hydrogen-bonding interactions with ASP228, ASP32, GLY34, and PHE108. π-π stacking was observed with TYR71 and hydrophobic interactions were seen with PHR108 and TYR71 (Fig. 3d).
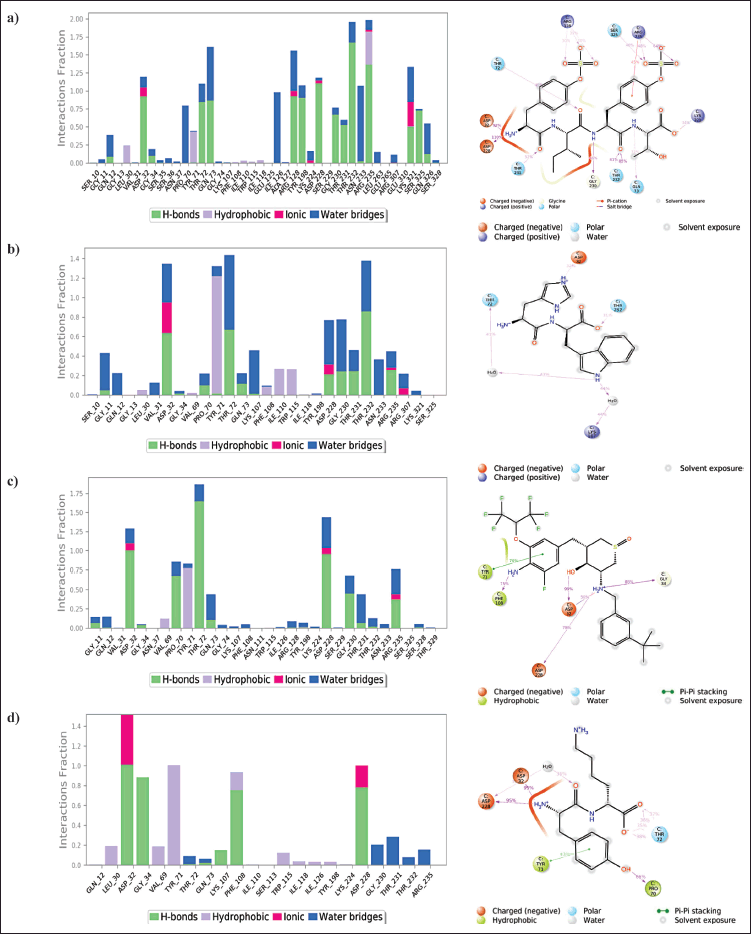 | Figure 3. Histogram of protein-ligand interactions. (a) Interaction of ZINC000150351431 with various BACE 1 protein residues (b) Interaction of ZINC000014945921 with various BACE 1 protein residues(c) Interaction of ZINC000069488328 with various BACE 1 protein residues (d) Interaction of CHEMBL2425609 (co-crystallized ligand) with various BACE 1 protein residues. The percentage of simulation time that a specific contact is maintained is represented by the value less than 1 in the stacked bar chart. A value greater than 1.0 implies that the same protein residue interacts with the ligand many times. [Click here to view] |
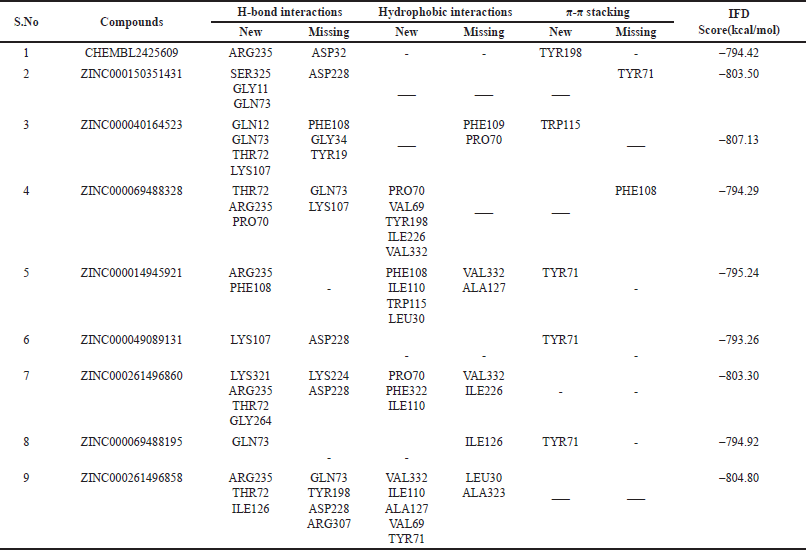 | Table 5. New and missing interactions of the top nine hits during induced fit docking in comparison to XP docking interactions and their IFD scores. [Click here to view] |
P-RMSF (Protein root mean square fluctuations) was utilized to visualize the fluctuations of each protein residue across the simulated time period. The protein amino acid residue that fluctuates more is represented by higher peaks. For complex 1, the most fluctuating residues were observed to be ALA313 with an RMSF value of 2.58 Å, GLN73 with RMSF value of 2.55 Å and GLY273 with RMSF value of 2.56 Å. RMSF of the remaining protein was found within the range of 1.02 Å to 2.58Å (Fig. 4a). For complex 2, the most fluctuating residues were observed to be VAL312 with RMSF value of 3.42 Å, SER46 with RMSF value of 3.00 Å and ALA157 with RMSF value of 3.00 Å. RMSF values of remaining protein was found within the range of 0.67 Å–3.42 Å (Fig. 4b). For complex 3, the most volatile residues were observed to be SER46 with RMSF value of 2.32 Å and ALA313 with RMSF value of 2.40 Å. RMSF volatility for remaining protein was found within the range of 0.56 Å–3.32 Å (Fig. 4c). For complex 4, the most volatile residues were observed to be THR314 with RMSF value of 4.06 Å, ASP311 with RMSF value of 2.89 Å and LEU167. RMSF volatility for remaining protein was found within the range of 0.60 Å–4.06 Å (Fig. 4d).
Ligand root mean square fluctuation (L-RMSF) could reveal how ligand fragments interact with proteins and have an entropic function in the binding process. For complex 1, O groups at position 34 and position 35 of the ring B of the structure were found to be the most volatile with an RMSF value of 4.43 Å and 4.37 Å, respectively. Other major fluctuations were seen with O atom at position 36 of ring B with an RMSF value of 4.01 Å. Overall the RMSF value of complex 1 with respect to protein ligand complex was observed within the range of 0.94 Å–4.43 Å (Fig. 5a). For complex 2, major volatility was seen with C alkene (position 7) of the imidazolinium ring with an RMSF value of 3.64 Å and with NH ion (position 6) of the imidazolinium ring with an RMSF value of 3.59 Å. Overall the RMSF value of complex 2 with respect to protein ligand complex was observed within the range of 2.14 Å–3.64 Å (Fig. 5b). For complex 3 major volatility was seen with NH group at position 1 with an RMSF value of 6.73 Å and with C atoms at positions 2, 3 and 4 with RMSF values of 5.78 Å, 4.88 Å and 3.80 Å, respectively. Overall, the RMSF value of complex 2 with respect to protein ligand complex was observed within the range of 0.67 Å–6.73 Å (Fig. 5c). For co-crystallized ligand—BACE 1, complex major volatility was seen with F (fluorine) atoms at position 1, 3, 4 with RMSF values of 2.29 Å, 2.34 Å, 2.15 Å, respectively. Overall, the RMSF value of co-crystallized ligand—BACE 1 complex with respect to protein ligand complex was observed within the range of 0.53 Å–2.36 Å (Fig. 5d). After a thorough analysis it was found out that aromatic ring, amine, and ketone groups were important structural features required for good interactions with the protein complex.
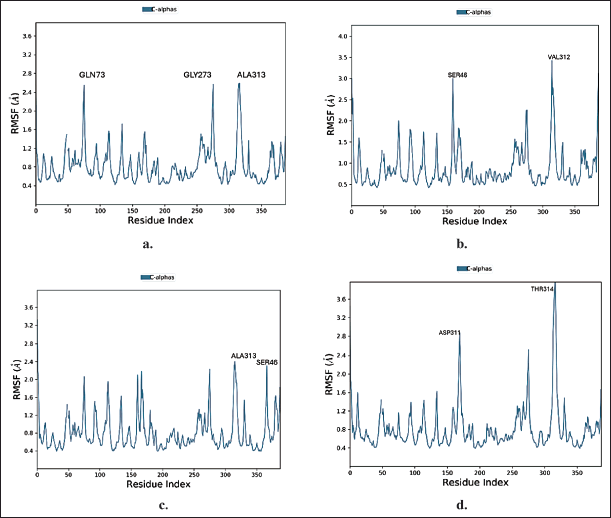 | Figure 4. Protein RMSF plot of a) ZINC000150351431 b) ZINC000014945921 c) ZINC000069488328 d) CHEMBL2425609 (co-crystallized ligand) during MD simulation. [Click here to view] |
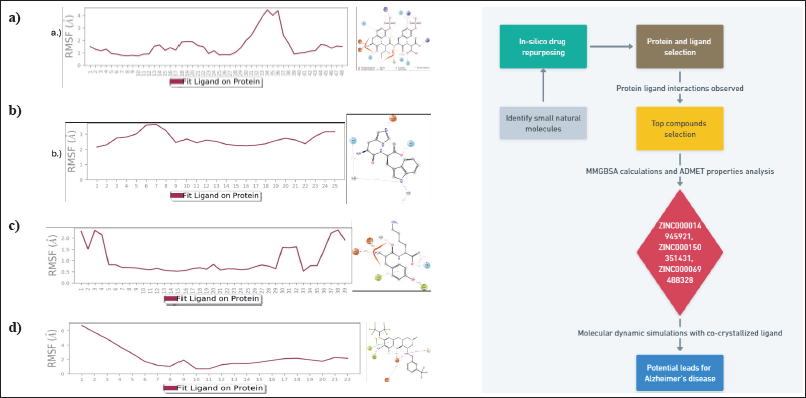 | Figure 5. Ligand RMSF plot of a) ZINC000150351431b) ZINC000014945921c) ZINC000069488328 d) CHEMBL2425609 (cocrystallised ligand) during MD simulation. [Click here to view] |
CONCLUSION
In the present work, rational approach receptor-based virtual screening was employed on the target (4LXM) to identify potential BACE 1 inhibitors. A library of natural products was prepared from ZINC 15 database. The hits were further analysed and ranked by using docking score, binding energy, fitness score, ADMET parameters and MD simulations. Thus, from these studies we suggest that ZINC000150351431 and ZINC000069488328 can act as possible leads for the treatment of AD. ZINC000014945921 comparatively was found to be less stable during MD studies as evident from RMSD plot data. However, the compounds need to be subjected to in vitro and in vivo studies to further strengthen our results.
ACKNOWLEDGEMENT
The authors would like to acknowledge Maipal College of Pharmaceutical Sciences for the computational facilities for carrying out this work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Association A. (2018) Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018;14:367–429. CrossRef
2. Kumar A, Srivastava G, Srivastava S, Verma S, Negi AS, Sharma A. Investigation of naphthofuran moiety as potential dual inhibitor against BACE-1 and GSK-3β: molecular dynamics simulations, binding energy, and network analysis to identify first-in-class dual inhibitors against Alzheimer’s disease. J Mol Model. 2017 Aug;23(8):239. CrossRef
3. Graham WV, Bonito-Oliva A, Sakmar TP. Update on Alzheimer’s disease therapy and prevention strategies. Annu Rev Med. 2017;68:413–30. CrossRef
4. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019, Alzheimer’s Dement. Transl Res Clin Interv. 2019;5:272–93. CrossRef
5. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment. [cited 2024 March 30].
6. Zhu C, Fu S, Chen Y, Li L, Mao R, Wang J, et al. Advances in drug therapy for Alzheime’s disease. Curr Med Sci. 2020;40:999–1008. CrossRef
7. Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS b-Amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8(363):1–14. CrossRef
8. Imbimbo BP, Watling M, Imbimbo BP, Watling M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs. 2019;28(11):967–75. CrossRef
9. Rueeger H, Lueoend R, Machauer R, Veenstra SJ, Jacobson LH, Staufenbiel M, et al. Discovery of cyclic sulfoxide hydroxyethylamines as potent and selective β-site APP-cleaving enzyme 1 (BACE1) inhibitors: structure based design and in vivo reduction of amyloid β-peptides, J Med Chem. 2012;55(7):3364–86. CrossRef
10. Joseph OA, Babatomiwa K, Niyi A, Olaposi O, Olumide I. Molecular docking and 3D qsar studies of C000000956 as a potent inhibitor of bace-1. Drug Res. 2019 ;69(08):451–7. CrossRef
11. Vassar R, Cole S. The basic biology of BACE1: a key therapeutic target for Alzheimers Disease. Curr Genomics. 2008;8(8):509–30. CrossRef
12. Koelsch G. BACE1 Function and inhibition: implications of intervention in the amyloid pathway of Alzheimer’s disease pathology. Molecules. 2017;22:1–20. CrossRef
13. Sorokina M, Steinbeck C. Review on natural products databases: where to find data in 2020. J Cheminform. 2020;12:1–51. CrossRef
14. Park JK. A review of computational drug repurposing. Transl Clinic Pharmacol. 2019;27(2):59–63. CrossRef
15. Atanasov AG, Zotchev SB, Dirsch VM International Natural Product Sciences Taskforce; Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20:200–16. CrossRef
16. Verma R, Boshoff HIM, Arora K, Bairy I, Tiwari M, Bhat VG, et al. Synthesis, evaluation, molecular docking, and molecular dynamics studies of novel N-(4-[pyridin-2-yloxy]benzyl)arylamine derivatives as potential antitubercular agents. Drug Dev Res. 2020;81(3):315–28. CrossRef
17. Choudhary MI, Shaikh M, Tul-Wahab A, ur-Rahman A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One. 2020 Jul 24;15(7):e0235030. CrossRef
18. Kumar S, Chowdhury S, Kumar S. In silico repurposing of antipsychotic drugs for Alzheimer’s disease. BMC Neurosci. 2017;18:1–16. CrossRef
19. Sundar S, Thangamani L, Manivel G, Kumar P, Piramanayagam. Molecular docking, molecular dynamics and MM/PBSA studies of FDA approved drugs for protein kinase a of Mycobacterium tuberculosis; application insights of drug repurposing. Inform Med.Unlock. 2019;16:100210. CrossRef
20. Parasuraman S, Raveendran R, Vijayakumar B, Velmurugan D, Balamurugan S. Molecular docking and ex vivo pharmacological evaluation of constituents of the leaves of Cleistanthus collinus (Roxb.) (Euphorbiaceae). Indian J Pharmacol. 2012;44(2):197–203. CrossRef
21. Maria LB, Nunzio I, Laura DL, Alba C. Induced-fit docking approach provides insight into the binding mode and mechanism of action of HIV-1 integrase inhibitors. Chem Med Chem. 2009;4:1446–56.
22. Veenstra SJ, Rueeger H, Voegtle M, Lueoend R, Holzer P, Hurth K, et al. Discovery of amino-1,4-oxazines as potent BACE-1 inhibitors. Bioorgan Med Chem Lett. 2018;28(12):2195–200. CrossRef
23. Chintha C, Carlesso A, Gorman AM, Samali A, Eriksson LA. Molecular modeling provides a structural basis for PERK inhibitor selectivity towards RIPK1. RSC Adv. 2020;10(1):367–75.
24. Paris D, Mathura V, Ait-Ghezala G, Beaulieu-Abdelahad D, Patel N, Bachmeier C, et al. Flavonoids lower Alzheimer’s Aβ production via an NFκB dependentmechanism. Bioinformation. 2011;6(6):229–36. CrossRef
25. Moussa-Pacha NM, Abdin SM, Omar HA, Alniss H, Al-Tel TH. BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med Res Rev. 2020;40(1):339–84. CrossRef
26. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17.
27. Iwaloye O, Elekofehinti O, Momoh AI, Babatomiwa K, Ariyo EO. In silico molecular studies of natural compounds as possible anti-Alzheimer’s agents: ligand-based design. Netw Model Anal Heal Inform Bioinform. 2020;9(54):1–14. CrossRef