
INTRODUCTION
Cancer statistics
Cancer is widely recognized as a prominent contributor to global mortality, resulting in approximately 10 million fatalities and 18.1 million confirmed cases worldwide, as reported by the World Health Organization [1]. The aberrant proliferation of cells, resulting from genetic and epigenetic changes, can lead to the spread of these abnormal cells throughout the body, migrating from one area to another [2,3].
The identification of the optimal therapy is hindered by several factors, such as the unique characteristics of each cancer type, the diverse reactions to treatment, and notably the absence of selectivity, which hinders the ability to target cancerous cells exclusively while sparing healthy tissues. As a result, there is a growing need to discover a new reliable target for cancer treatment [4].
Glyoxalase system
The glyoxalase system has long been a subject of attention as a possible target for the development of new anticancer drugs. The glyoxalase complex consists of two enzymes: glyoxalase I (Glo-I) and glyoxalase II (Glo-II). Both enzymes function as detoxifying agents for deleterious byproducts, such as methylglyoxal (MG), which are generated during the body’s regular metabolic processes, such as glycolysis [5–7]. MG and glutathione (GSH) undergo a nonenzymatic reaction to produce a hemi-thioacetal substrate. This substrate is then catalyzed by Glo-I to make S, D-lactoylglutathione, which serves as a substrate for Glo-II. Glo-II hydrolyzes the thioester into the less hazardous D-lactic acid (Fig. 1) [5,8]. Glo-I is overexpressed in these cells because most tumor cells have enhanced metabolic activity, which raises intracellular hazardous compounds (including MG) [9,10]. Furthermore, there is a correlation between Glo-I and chemotherapy in combating multidrug resistance in cancer [11]. The results identified the Glo-I enzyme as a promising target for the development of anticancer medicines [12].
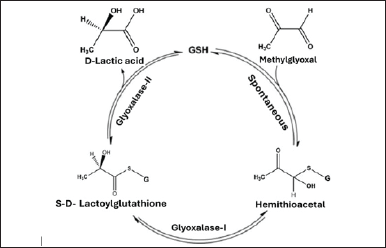 | Figure 1. Illustration of the process of detoxifying MG through the glyoxalase system. [Click here to view] |
Active site geometry
The majority of tumor cells demonstrate augmented glycolytic activity, resulting in higher levels of hazardous compounds such as MG within the cells [13]. Tumor cells enhance the expression and activity of the glyoxalase system, specifically Glo-I, to lower the intracellular levels of hazardous metabolites. This is done to prevent cellular growth arrest and ensure continued growth [14].
Glo-I is a protein composed of two subunits with a molecular mass of 42 kDa. It is encoded by the human Glo-I gene and belongs to the category of enzymes that require zinc ions for their activity. An analysis was conducted on the active site to determine the essential characteristics needed for optimal complementarity between the enzyme and inhibitor [15]. Three essential criteria were identified as crucial: a deeply hydrophobic cavity capable of accommodating a hydrophobic entity, a region that binds zinc and forms a complex with Zn2+, and a positively charged entrance to the active site that can electrostatically bind to negatively charged groups in inhibitors (Figs. 2 and 3) [16,17].
The active site entryway is encircled by the amino acids Lys150, Arg37, Arg122, and Lys156. Once they acquire a positive charge, they will engage in electrostatic interactions with the inhibitors. Simultaneously, the zinc atom located at the core of the active site interacts with Glu172B, Gln33A, His126B, and Glu99A, resulting in the formation of various coordination geometries such as octahedral, square pyramidal, or trigonal bipyramidal. Finally, there is a significant hydrophobic cavity located deep within the active site that cannot be reached by solvents. This cavity is delineated by the amino acids Leu92, Phe71, Met179, Leu160, Leu69, and Phe62 [18–21].
Study rationale
Tumors have a high demand for glucose to fuel their rapid growth, leading to the generation of large amounts of the poisonous MG. The Glo-I enzyme plays a crucial function in safeguarding cells against the harmful effects of MG. Its increased expression in tumor cells has been identified as a key component of their survival strategy [22, 23]. This survival strategy has been observed in various types of tumors, including breast cancer, gastric cancer, colon cancer, and bladder cancer [8, 24]. Furthermore, there is a correlation between Glo-I and chemotherapy in relation to the multidrug resistance of cancer. The results of these studies have identified the Glo-I enzyme as a promising candidate for the creation of new and innovative anticancer drugs functions as a Glo-I inhibitor, while there are two categories of Glo-I inhibitors: GSH-based inhibitors and non GSH-based inhibitors [12, 25–27].
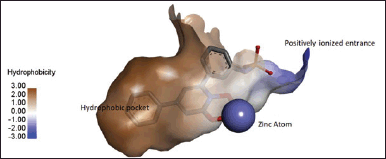 | Figure 2. Displays the active site of Glo-I, illustrating the key locations for inhibitor bindings in 3D structure (N-Hydroxypyridone co-crystalized ligand bound in bidentate geometry to zinc atom at the active site from PDB 3W0T). [Click here to view] |
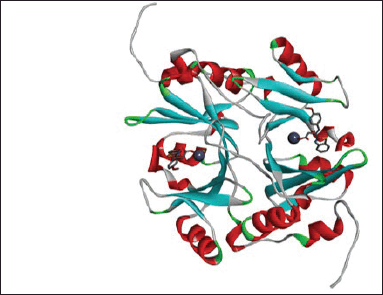 | Figure 3. Cartoon representation of Glo-I, using the PDB code 7wt2 and a resolution of 2 Å (TLSC702 co-crystalized ligand bound to zinc atom at the two active sites from PDB 7WT0). [Click here to view] |
Computer aided drug design (CADD) utilizes the three-dimensional structure and shape of the target binding site to assess and create novel ligands and drugs by considering their interactions [28]. The subcategories of CADD include Structure-based drug design (SBDD) and ligand-based drug design (LBDD). SBDD focuses on identifying a suitable therapeutic target and acquiring its structural information. On the other hand, LBDD is employed when the three-dimensional structure of the protein is not accessible, and the data is derived from a set of active ligands that interact with a specific target, such as an enzyme or a receptor [29,30]. LBDD has emerged as a crucial computational method for drug development in cases where a macromolecular target structure is unavailable. Usually, this approach entails determining common chemical properties based on the three-dimensional arrangements of a set of known ligands that are essential for interacting with a particular macromolecular target [31]. Both techniques were employed in the current study. The LBDD was employed to generate reliable pharmacophores, which were subsequently utilized to retrieve corresponding chemicals from a commercial database. Subsequently, the candidate compounds obtained via LBDD were submitted to a prioritizing process using SBDD techniques, including molecular docking and estimations of “TBE.” Afterward, the candidate compounds were subjected to in vitro testing against the Glo-I enzyme.
MATERIALS AND METHODS
Compound preparation
The chemicals selected for this study were sketched using ChemBioDraw Ultra® and subsequently loaded into discovery studio (DS). The corresponding standards were used to convert them into 3D structures. The minimize ligand protocol in DS was utilized to generate 3D structures using the default parameters.
Training set selection
We selected 12 chemicals as a training set for our investigation. Each compound within the active group has been assigned a principal value of 2, with a maximum omitted feature value of 0. Concurrently, the principal value of the inactive compounds was assigned as 0, while the maximum omitted feature value was set to 2. Subsequently, distinct files were generated for the active test set, inactive test set, and the compounds of the training set.
Ligand based pharmacophore generation
The common feature pharmacophore protocol was employed to construct pharmacophore models within this study. The protocol involved analyzing a series of ligands to uncover shared characteristics.
There are two approaches that can be used in DS to build 3D conformations: the on-the-fly method and the pre-numerating method. The on-the-fly approach is used to produce conformations while moving through the pharmacophore modeling process. Alternatively, the pre-numerating approach involves creating many confirmations for each individual molecule beforehand and storing them in a database. This process is accomplished by generating a confirmation protocol. The conformers of the compounds of interest were created using the on-the-fly technique in our work [32]. Furthermore, the option of “best conformer generation” was employed, with the maximum number of conformer generations per molecule set to the default value of 255. The energy threshold was modified to 10 kcal/mol.
Validation of the generated pharmacophores
The validation of the generated pharmacophores was conducted to ensure that our model can accurately differentiate between active and inactive compounds, and to confirm its reliability in identifying potential hits with diverse chemical structures from the ASINEX® database [33].
The pharmacophore model built by the hip-hop-refine module of DS was evaluated using a test set of 56 chemicals for locating the active molecules. The activity was classified based on the IC50 value, with compounds having a value greater than 50 µM being deemed inactive, while compounds with a value less than 50 µM were considered active. The test set was employed to validate the pharmacophore models and assess their capacity to differentiate between inactive drugs and active ones. The validation step was included in the common feature pharmacophore creation methodology. Our investigation utilized the pharmacophore model that yielded the most favorable validation findings.
The evaluation method was conducted by analyzing receiver operating characteristic (ROC) curves and computing the corresponding statistical descriptors, following the steps outlined below.
ROC curves analysis
The ROC curves were employed as a validation approach to assess the pharmacophore’s capacity to differentiate between active and inactive compounds. The x-axis represents the false positive rate, which is calculated as 1 minus the specificity. On the other hand, the y-axis represents the true positive rate, also known as sensitivity. The ROC area is a numerical metric that quantifies the predictive capabilities of a model. It ranges from 0.5, indicating random prediction, to 1.0, indicating outstanding prediction [34].
We created ROC curves for the models utilizing internal validation of the common feature pharmacophore-generating procedure. The created pharmacophore models were analyzed using the ROC curve to show their sensitivity and specificity. Equation 1 defines sensitivity as the ability of the model to accurately detect active chemicals in the test set and differentiate them from inactive compounds. Specificity, as defined by Equation 2, measures the model’s accuracy in correctly identifying inactive chemicals in the test set.
Sensitivity of ROC curve analysis, TP refers to the number of true positive compounds, whereas FN refers to the number of false negative compounds.
Specificity of ROC curve analysis, TN refers to the number of true negative compounds, whereas FP refers to the number of false positive compounds.
Calculation of statistical descriptors
The Güner-Henry (GH) evaluation method was successfully utilized on a dataset that included both active and inactive compounds. The efficacy of this scoring method has been demonstrated in assessing diverse facets, including model selectivity (determining the optimal model), accuracy in detecting active compounds, and retrieval of active compounds. Furthermore, the GH scoring approach has demonstrated encouraging outcomes in the field of database mining, aiding in the measurement of model selectivity and the extent of coverage in the activity space. The GH analysis consisted of computing several parameters, as demonstrated in (3a) the percentage of active compounds (%A), which indicates the extent of the model’s activity space coverage, (3b) the percentage yield (%Y), which quantifies the model’s selectivity, (3c) the enrichment factor (E), and (d) the GH scores (Eq. 3).
Equation (3a) %A: the percentage of active compounds, (3b) %Y: the percentage yield, (3c) E: the enrichment factor, and (d) the GH scores.
The values of these variables were derived from the data obtained from the drug database, including the total number of compounds (D), the number of active compounds (A), the number of correctly retrieved active compounds by the model (Ha), and the total number of compounds retrieved by the model as hits (Ht). A GH score ranging from 0.7 to 0.8 indicates a highly effective model [35,36].
Selection of the final pharmacophore
After evaluating the validation results using the quality of ROC curve values and GH score analysis, the top three pharmacophores were selected for searching the 3D database.
Virtual screening process
The 3D pharmacophores that were created were utilized to retrieve hits from the ASINEX® database. The process began by constructing a 3D database, utilizing predetermined parameters. We set the confirmation parameter to the “best” option to generate the confirmation. The compounds have been converted into their corresponding 3D conformations using the “build 3D database” technique. The chosen pharmacophores underwent screening using the Search 3D database technique to find possible inhibitors of Glo-I. The best align method parameter was used, while the other parameters remained in their default state.
Filtration of the retrieved hits
The obtained results were subjected to filtration based on many criteria, such as Lipinski’s rule of five and Veber’s rule for drug-like properties, utilizing the filtration protocols developed by Lipinski and Veber. Subsequently, the hits that satisfied all three pharmacophores were chosen, and any duplicates originating from distinct pharmacophores were removed.
Compounds preparation
Before initiating the docking procedure, the final group of refined hits underwent preparation using the “Prepare Ligand” protocol. The preparation process utilized the default parameters, with the exception of the tautomer enumeration, which was explicitly configured to utilize the canonical tautomer.
CDOCKER docking protocol
The CDOCKER algorithm, which is based on the CHARMM forcefield, is a molecular docking technique that uses a grid-based approach. The CDOCKER algorithm generated multiple binding conformations for each molecule. For each of the 10 poses of each ligand, two energies were computed: the CDOCKER energy, which accounts for both the interaction energy between the protein and ligand, as well as the internal strain energy of the ligand. Furthermore, the CDOCKER interaction energy quantifies the energy linked to nonbonded interactions between the protein and the ligand. The parameters were maintained in their default configuration. Subsequently, 10 poses were constructed for every ligand, and the related energies were computed for each pose. To verify the reliability of the docking methodology being used, it is recommended to first perform this process with a ligand-enzyme complex (provided one is available). This will allow for the assessment of the algorithm’s ability to accurately identify the binding site. This step is performed by first extracting the ligands and then redocking them into the active site (5ZO and HPJ).
Calculation of the binding energy
Before estimating the binding energy, we employed the “in situ ligand minimization” parameter, which we subsequently converted into the adopted basis Newton Raphson (NR) technique with default steps. The ligand-receptor binding free energies are calculated using the “calculate binding energies” parameters in DS. The default settings are used, with some specific changes made for the ligand conformation and solvent model. The ligand conformation parameter is set to “true” and the solvent model is changed to the implicit solvent model, specifically Poisson Boltzmann with the nonpolar surface area (PBSA) and the “Best” setting for the ligand conformational parameters [37]. Moreover, this parameter facilitates the calculation of the mean binding energy for a set of related positions, taking into account the loss in conformational entropy and the energy of the bounded ligand [38]. The calculation of the binding energy is carried out using the following equation (Eq. 4).
Energy Binding = Energy Complex – (Energy Ligand + Energy Receptor) (4)
Equation for the computation of the binding energy.
Biological evaluation of the selected compounds
To evaluate the ability of the selected compounds to inhibit human Glo-I, we performed an in vitro Glo-I experiment utilizing human recombinant Glo-I (rhGlo-I) [39]. The Glo-I assay utilizes a spectrophotometric technique to measure the increase in absorbance at 240 nm, which serves as an indicator of the synthesis of S-D-lactoylglutathione.
The inhibitory effects of the selected compounds were determined by assessing their biological activities using an in vitro test on rhGlo-I, following the manufacturer’s protocol (R&D Systems, Inc., Minneapolis, MN). The rhGlo-I enzyme was reconstituted by dissolving it in sterile and deionized water to achieve a concentration of 0.5 mg/mL. The reconstituted enzyme was then stored at a temperature of −70 °C and thawed on the day of the test. The chemicals were dissolved in dimethyl sulfoxide (DMSO) to create a stock solution with a concentration of 10 mM. The test buffer was prepared by mixing a 0.1 M sodium phosphate dibasic solution with a 0.1 M sodium phosphate monobasic solution, resulting in a pH range of 7.0 to 7.2. The test compounds were dissolved in DMSO to form a stock solution with a concentration of 10 mM. The absorbance of the solution was then measured at a wavelength of 240 nm for a duration of 200 seconds at a temperature of 25°C.
The inhibitory effect of the chosen hits against Glo-I was assessed in vitro using rhGlo-I (recombinant human Glo-I) at positions [39]. To carry out this evaluation, three distinct experiments were conducted in triplicate for each chosen sample utilizing a plate reader, and the mean of these outcomes was computed. The IC50 values of all chosen hits were computed using GraphPad Prism 8 Figure 4.
Slope of kinetic mode.
Percentage of activity.
Percentage of inhibition.
RESULTS AND DISCUSSION
Glo-I enzyme inhibitors compilation
In this work, previously known Glo-I enzyme inhibitors with varying activity profiles were retrieved from the literature. This procedure yielded 59 active compounds, with an activity threshold of 50 µM. The biological assay used to determine the activities was identical in the majority of cases, indicating the accuracy of the pharmacophore model that will be later built. The activity ranged from 0.011 to 50 µM with various chemical scaffolds. To generate a common pharmacophore, nine decoys with activity greater than 50 µM were selected from literature and in-house nonactive compounds.
ChemDraw® 12.0 was used to sketch the obtained compounds from the literature, and the structures were confirmed by a third party. The 3D coordinates of the compounds were obtained by transferring them to DS 2022, which was subsequently prepared and minimized using the default parameters while eliminating isomerization and tautomerization steps. This approach yielded the same amount of chemicals, which will subsequently be categorized. The 59 compounds were allocated to be active within the DS by adding two columns: “Principal” and “MaxOmitFeat,” with “Principal” set to 2 and “MaxOmitFeat” set to 0.
In contrast, the inactive compounds were assigned a value of 0 for “Principal” and 2 for “MaxOmitFeat.” The next stage was to unravel the features that will be employed in the pharmacophore generation step using the “feature mapping” procedure. This stage yielded five features: “zinc binder, negative ionizable, H-bond donor, H-bond acceptor, ring aromatics, and hydrophobic.”
Common feature pharmacophore generation
This stage involves selecting a training set in which the common feature pharmacophore is utilized to generate a sequence of pharmacophores with varying features. The training set (see Table 1) was chosen to include 12 compounds, with 10 active and two inactive. The compounds were chosen based on their broad activity range and structural dissimilarity, with the remaining compounds serving as the test set.
The next step was to run “common feature pharmacophore generation.” The settings utilized in this technique were tailored to our specific needs. The energy threshold was set to 10 kcal/mol, which is known to provide a realistic structure during the pharmacophore generation process. The minimum inter-feature distance of 2Å yielded the best results based on the ROC curve. The fitting method was specified to be flexible, with “true” for the validation phase. This will verify the legitimacy of the obtained pharmacophores. The minimum number of features has been set to four, with a maximum of five features. The number of leads that may be missed was set to one because it produced the best ROC results.
This procedure yielded 10 pharmacophores (Table 2). Three pharmacophores were chosen for the screening process, and the results were validated (Table 3). These pharmacophores were chosen based on ROC curve analysis and were classified as “good” with values ranging from 0.80 to 0.90, while the remaining pharmacophores were classified as “fair.” Pharmacophore 1 has a ROC curve value of 0.83 and has produced two HBAs, one HBD, and one ring aromatic. Pharmacophore 2 has a ROC value of 0.82 and shares the same properties as pharmacophore 1. Pharmacophore 4 had an ROC of 0.83, and the same features were exhibited (Fig. 5).
Because this enzyme has a zinc feature in its active site that serves both structurally and catalytically, we began a process of manually replacing one of the features with a “zinc binder.” This technique was carried out by mapping the co-crystalized azaindole hydroxypyridone compound onto the resulting pharmacophores and replacing the hydroxamic acid mapped feature with zinc binder. One of the hydrogen bond acceptors (HBAs) was substituted (Fig. 6), and the process was repeated for the remaining three pharmacophores. This method was critical for our research because the major goal was to discover zinc-binding groups rather than HBA. Once completed, the pharmacophore will only detect the zinc-binding functional group in the search database.
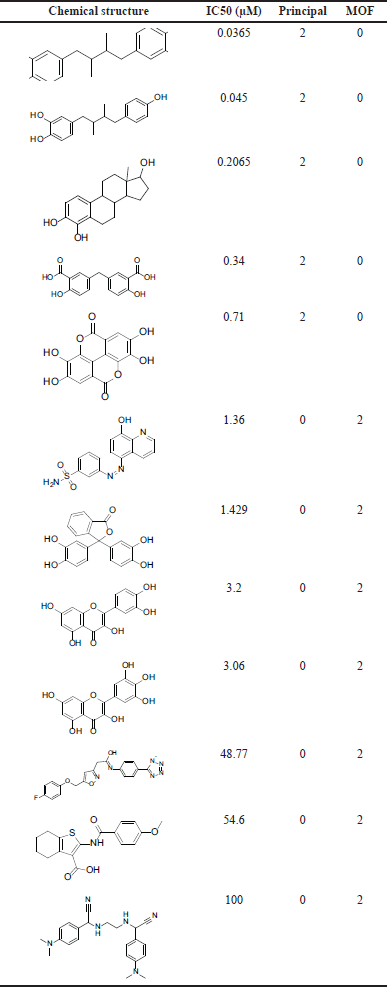 | Table 1. The training set in our study, MOF: MaxOmitFeat. [Click here to view] |
Gunner–Henry score method
The pharmacophore model was further validated using the well-known Gunner–Henry score (GH). This procedure is used to assess the model selection precision of pharmacophore hits and to distinguish between actives and inactives. When the GH score is one, the goodness of hits is optimal, whereas a zero indicates poor outcomes. The GH value for the first pharmacophore was 0.92, the second pharmacophore was 0.91, and pharmacophore 4 had the highest value of 0.93. These findings have bolstered our efforts and lent credence to our work. Normally, a value of 0.7–0.8 suggests a solid correlation model.
Database search
The Asinex® Commercial Database of about half a million compounds was searched to find matches that meet the three pharmacophore models. This method began with the database being prepared ahead of time to simplify the extraction process. The database was created using the “build 3D database” protocol in DS®, which generated and saved the 3D coordinates and conformers for later usage. The first pharmacophore yielded 2,070 hits, the second 2,315, and the third 2,790. The hits that are common on the three pharmacophores were picked for the next filtering phase utilizing stringent hit compound selection and minimization without personal prejudice. To increase the validity of our compounds and ensure that these hits have advantageous orally absorbable properties, a filtration step using Lipinski’s rule of five and Veber’s rule was performed. These steps yielded 368 hits that demonstrated all pharmacophore mapping.
Docking and total binding energy (TBE) study
The 368 hits obtained were further filtered by subjecting these compounds to a docking study utilizing the DS CDOCKER protocol. The hits were generated using the “prepare ligands” technique, which prevented any isomerization while permitting rule-based ionization and canonical tautomerization, yielding 522 molecules for the docking stage. The docking procedure was performed on two Glo-I Protein Data Bank (PDB) entries: 3VW9 and 7WT2. Our study group had already prepared, solved, and reduced these two enzymes. The results of this stage were further used to do “in-situ” minimization and calculate the “TBE.” This approach will help us as researchers select the final molecules with biological activity.
Final selection process
The selection criteria were a combination of experimental data and specific conditions relating to our prior knowledge of active site shape. The first selection stage was based on “CDOCKER interaction energy” and TBE for the two enzymes with the best results (no positive TBE and low CDOCKER). This stage reduced the number of compounds selected for visual evaluation from 368 to 59. The final selection process was carried out using the available in silico results, with significant assistance from the researchers as experienced experts in this enzyme. The interference procedure was necessary at this stage to prevent black box behavior, in which what the computer generated is taken for granted. It is generally understood that the input to in silico work is determined by the quality of the structures placed or available. As a result, the researchers’ experience could aid in mitigating prejudice that may arise during in silico study. A total of 15 compounds were chosen from the 59 compounds (Table 4). These compounds are distinguished by their ability to hold a variety of chemical scaffolds with different 3D geometry.
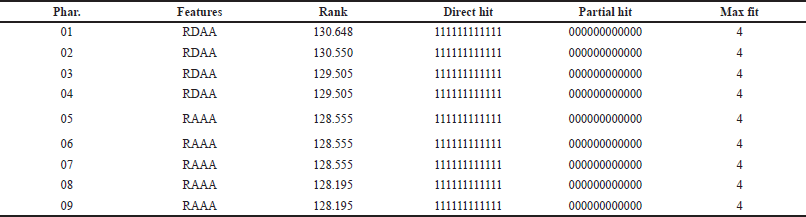 | Table 2. The value of the top 10 pharmacophores hypothesis obtained from HipHopRefine algorithm of our study. [Click here to view] |
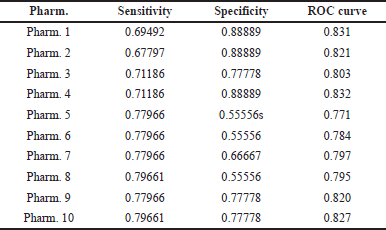 | Table 3. The validation results of our study. [Click here to view] |
Investigating the structures of the selected compounds reveals that they all contain a carboxylic acid group, which may indicate the importance of this group in chelating the zinc atom at the active site. In most cases, this carboxylic acid is immediately linked to the aromatic ring, which is required to fill the hydrophobic region adjacent to the zinc atom at the active site. This hydrophobic pocket can hold up to two aromatic rings. The presence of carboxylic acid can also be attributed to the existence of a crucial property of the active site, namely the very positive entry. In most docking situations, the carboxylic acid has been directed away from being a zinc binder and toward ionic interactions in this area. As a result, the zinc-binding moieties in these compounds could be converted into alternative zinc binder functions such as ketone, thione, amide, sulfide, and thiazolidinedione (negatively ionized). The selected chemicals were purchased and tested in vitro except for two compounds that were unavailable. It should be noted that the flexibility of the molecules was considered when selecting the potential compounds. The majority of the compounds were chosen to be semi-rigid with three-ring configurations.
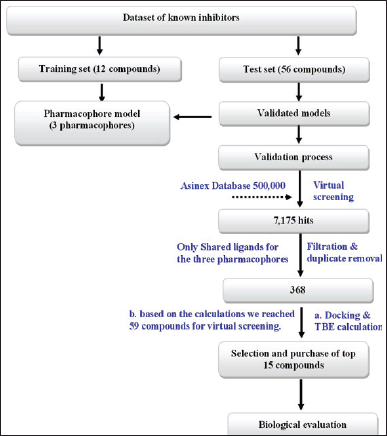 | Figure 4. Summarized steps of project methodology. Note: A “training set” typically consists of a selection of compounds that are previously known to have activity against the target. This set includes compounds with a wide range of activity levels, ranging from very active to inactive. The purpose of the training set is to guide the software in constructing a sequence of pharmacophores. The “test set” refers to a bigger collection of compounds that have already been identified as having a certain activity. We utilized this set to evaluate the effectiveness of the pharmacophore developed from the training set in differentiating these compounds as either active or inactive. If the prediction was accurate, we would consider the created pharmacophores for future experimentation. These pharmacophores will be utilized to identify possible inhibitors from commercial databases by extracting unknown chemicals. [Click here to view] |
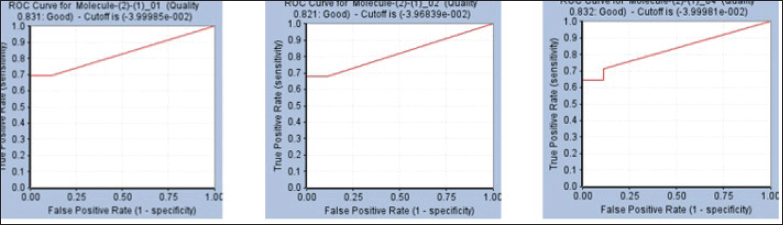 | Figure 5. ROC curves resulted from the generated pharmacophore models. [Click here to view] |
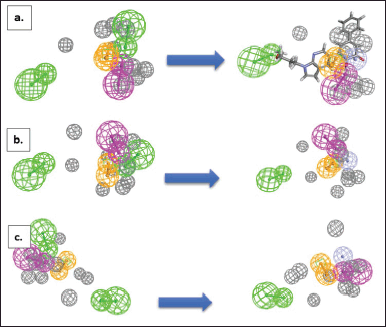 | Figure 6. The modified analogues of the pharmacophore feature (a) pharmacophore-1, (b) pharmacophore-2, (c) pharmacophore-4. The modifications were carried out by changing the HBAs (on the left) into zinc-binding feature (on the right, magenta sphere). The green color represents HBA, the brown color represents ring aromatic, and the purple color represents HBD. The grey color represents the exclusion sphere. [Click here to view] |
In-vitro biological assay
The inhibitory activity of the selected hits against rhGlo-I was determined in vitro using rhGlo-I [21]. Three successive tests were performed in triplicate on each of the selected hits using a plate reader, and the average of these results was calculated. GraphPad Prism 8 was used to calculate the IC50 values for all selected hits. The data are presented as a percentage of inhibition. This test was performed in-house in accordance with the R&D company’s procedures. Myricetin (3.06 µM) was employed as a positive control on customized UV-compatible plates. The wavelength of measurement was set to 240 nm, which is in the ultraviolet range. This method was performed three times to guarantee the test’s correctness, and a third party tested them to prevent any personal errors. Four of the 13 compounds demonstrated activity of less than 50 µM, with the highest activity at 2.79 µM. (Fig. 7) depicts the IC50 curves.
Active compounds analysis
BAS00323528, the most active chemical, inhibits Glo-I enzyme at a concentration of 2.79 µM in vitro. This molecule is composed of three primary structural blocks: the war head zinc binder thiazolidinedione ring, which is expected to have a negative charge at physiological pH. This negative charge will establish a coordination bond with a zinc atom at the active site. Because of the role of zinc atoms in enzyme mechanisms, this bond is regarded as the most important feature in reflecting the compound’s activity. Furthermore, this ring has a hydrophobic carbon skeleton along with a sulfur atom, which will generate hydrophobic interactions with the hydrophobic pocket next to the zinc atom. The salicylic acid moiety on the opposite side of this structure, which is likewise expected to have a negative charge (Fig. 8), was in charge of generating ionic connections with the positively ionized active site entrance. Lys156 is the primary basic amino acid responsible for binding to salicylic acid. The third structural element is the middle furan ring, which forms hydrophobic contacts with Met 65 and Met 157.
BAS02067456 was the second most active chemical, with an IC50 of 20.25 µM. This chemical contains a potential zinc chelating ring, 2-thioxothiazolidin-4-one, which is responsible for zinc chelation and hydrophobic interactions (Fig. 9). The ketone activity chelates the zinc atom, whilst the thione moiety engages in hydrophobic interactions. The adjacent hydrophobic pocket was optimally filled by a thiophene ring, which interacts hydrophobically with a variety of amino acids including Leu92, Cys60, Met183, and Met179. The salicylic acid moiety in the prior molecule likewise has similar ionic interactions with the active site entryway, particularly with Arg37.
BAS01854346 was the third most active chemical, with an IC50 of 26.2 µM. The zinc-binding moiety was a benzothiazole ring in which the sp2 hybridized nitrogen atom chelates the zinc atom, similar to the imidazole ring found in His amino acids. Furthermore, the sulfur atom in this ring structure is important for hydrophobic interactions. The benzonitrile functionality accommodates the hydrophobic moiety in a unique way, with the nitrile responsible for H-bonding with Cys60 and the aromatic ring performing hydrophobic interactions with Met179, Met157, and Phe62. The benzoic acid functional group may be capping the active site moiety through ionic interactions with Arg37 (Fig. 10).
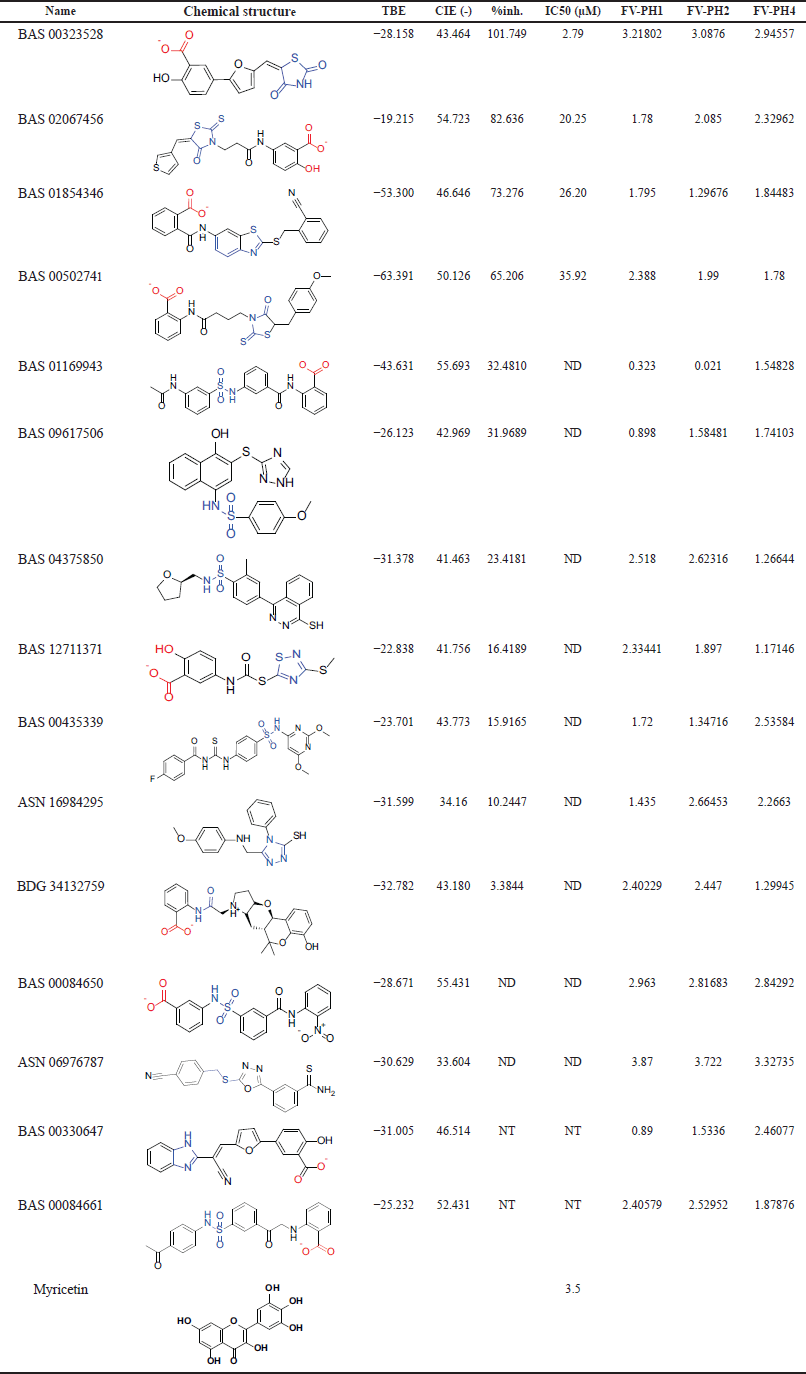 | Table 4. The selected 15 compounds for characterization. The blue-colored moiety is related to the zinc-binding group. The first four active compounds in addition to Myricetin are shown in Figure 7. TBE: TBE, CIE: CDOCKER interaction energy, %inh.: percentage of inhibition, ND: not determined, NT: not tested, FV-PH: Fit value of pharmacophore 1, 2, and 4. [Click here to view] |
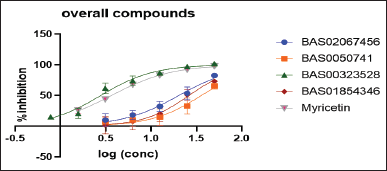 | Figure 7. Dose-response curves for the compounds (BAS02067456, BAS0050741, BAS00323528, BAS01854346, and Myricetin). [Click here to view] |
BAS00502741 was the next most active, having an IC50 of 35.29 µM. This chemical was discovered to be strikingly similar to BAS02067456. As expected, an identical binding pattern was seen when the 2-thioxothiazolidin-4-one performed zinc chelation, with the sulfide structure responsible for the chelation process. The methoxy bioisostere of the thiophene ring of this molecule occupied the hydrophobic pocket and interacted with Leu182, Met183, Ile88, and Cys90. The negatively ionized salicylic acid functionality demonstrated an ionic interaction with Lys156 at the active site entrance. Interestingly, when docked inside the active site, this molecule takes on a different orientation, with the salicylic acid exhibiting zinc chelation capabilities because the negative ion on the carboxylic acid is linked with zinc. The methoxy ring had ion-dipole interactions with the positively charged entrance (Fig. 11).
Concerning the inactive compounds, their presence warrants investigation and a clear explanation for their lack of activity is useful in avoiding them in the future. There are several typical explanations for the lack of activity, including the absence of an effective ROC pharmacophore, which will be reflected in the quality of compound selection. Furthermore, during the structure-based evaluation step, the available protein structures are expected to be less optimum due to a variety of factors such as crystallization quality and resolution, as well as the type of bound ligand.
For example, compound BAS00084650 is projected to have sulfonamide as its core zinc chelation component. It is predicted that this functional group was unable to chelate zinc because it was unable to generate an ionized form due to the lack of an Electron Withdrawing Group. So, the pKa value will be between 10 and 11. Compound ASN06976787 was assumed to be active because it comprises the oxdiazole ring as a zinc chelating moiety, with one of the ring’s nitrogens coordinating with the zinc atom. However, the biological assay yielded no enzyme inhibition. This could be attributable to the absence of a negatively charged moiety in this structure, as well as inappropriate orientation inside the active site. One cause could be the compound’s low solubility profile during test measurements, albeit this was not fully proven due to the small amounts employed in this assay. Given our prior knowledge that compounds BDG 34132759 would be inactive, this was purchased and tested to provide additional confirmation that positively ionized compounds will undoubtedly lack biological activity, which could be explained by their inability to cross the highly positively ionized active site entrance.
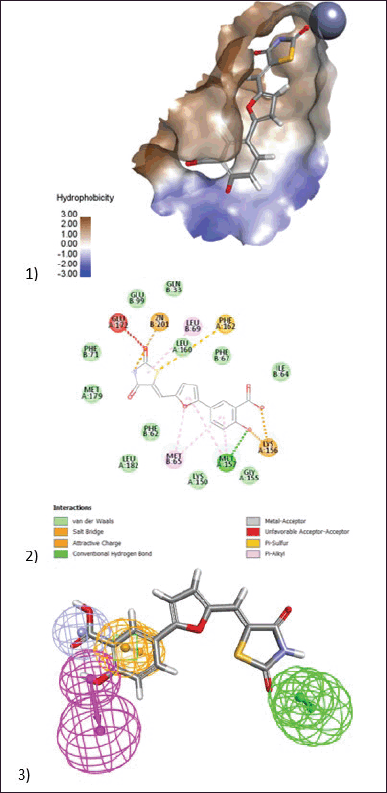 | Figure 8. (1) 3D model of docking of the compound (BAS00323528) in the active site of Glo-I enzyme. The hydrophobic surface was used to depict the binding area. (2) 2D model represents the interaction between the compound (BAS00323528) and the active site of the enzyme. (3) Elucidation of (BAS00323528) compound in stick pattern mapped to Pharmacophore-1. [Click here to view] |
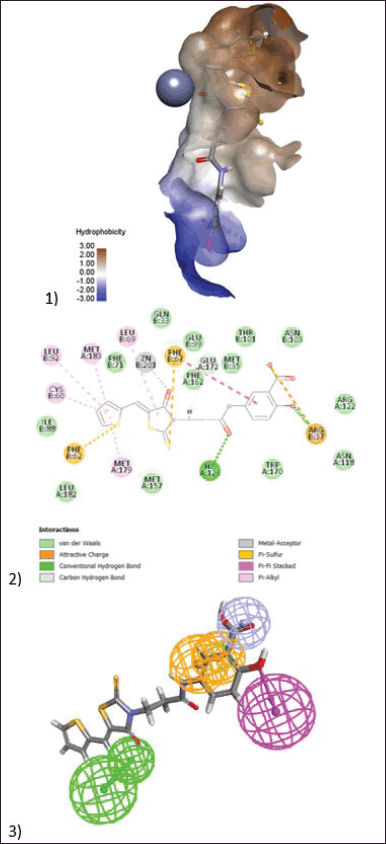 | Figure 9. (1) 3D model of docking of the compound (BAS02067456) in the active site of Glo-I enzyme. The hydrophobic surface was used to depict the binding area. (2) 2D model represent the interaction between the compound (BAS02067456) and the active site of the enzyme. (3) Elucidation of (BAS02067456) compound in stick pattern mapped to Pharmacophore-1. [Click here to view] |
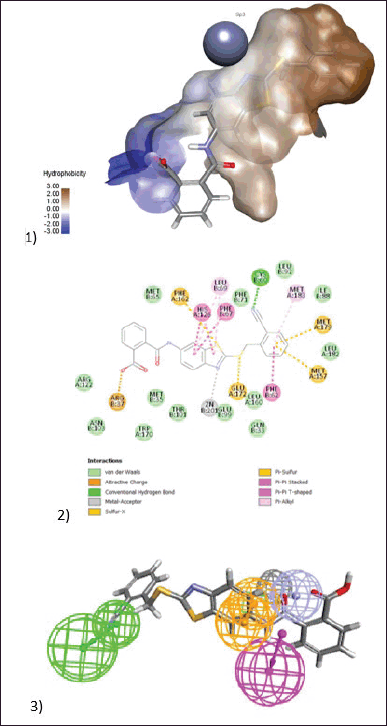 | Figure 10. (1) 3D model of docking of the compound (BAS01854346) in the active site of Glo-I enzyme. The hydrophobic surface was used to depict the binding area. (2) 2D model represents the interaction between the compound (BAS01854346) and the active site of the enzyme. (3) Elucidation of (BAS01854346) compound in stick pattern mapped to Pharmacophore-1. [Click here to view] |
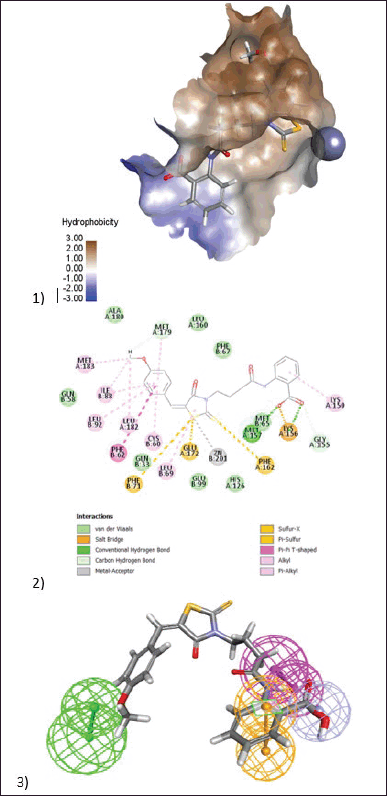 | Figure 11. (1) 3D diagram of (BAS00502741) in the active site of Glo-I enzyme. The hydrophobic surface was used to depict the binding area. (2) 2D model represents the interaction between the compound (BAS00502741) and the active site of the enzyme. (3) Elucidation of (BAS00502741) compound in stick pattern mapped to Pharmacophore-1. [Click here to view] |
CONCLUSION
The combination of Ligand Pharmacophore modeling and SBDD approaches is widely regarded as a reliable and potent approach for identifying novel inhibitors with high potential. In this study, we integrated both strategies to discover previously unknown chemical scaffolds that exhibit specific inhibitory activity against Glo-I. Three out of ten pharmacophore models have demonstrated high ROC quality and an exceptional Gunner–Henry score. The pharmacophores were employed to filter the ASINEX® database, which contains around 500,000 chemicals. Subsequently, the commonly occurring hits that matched the three pharmacophores were chosen and then submitted to a more rigorous filtration process, including Lipinski’s rule of five and Veber’s rule. The compounds that received approval were subjected to docking, and their overall binding energy was computed, which was subsequently utilized in the process of refinement. As a result, a total of 15 compounds with different chemical frameworks were chosen and acquired. Their biological efficacy was assessed in vitro against the Glo-I enzyme, yielding four active compounds. Among these, BAS00323528 exhibited the highest level of activity, with an IC50 value of 2.79 µM. The success rate of our model is commendable, with four out of thirteen compounds (excluding two compounds that were not available in the commercial collection) exhibiting action at a rate of 30%. Fortuitously, this occurrence of a hit rate is unprecedented within our research group and will facilitate the development of our future pharmacophore models. This endeavor will provide researchers with extensive opportunities to utilize various computer-aided drug design tools to develop novel inhibitors by suing both active and inactive molecules. The active compounds can serve as a reference for conducting in silico search operations and as a basis for synthesizing new inhibitors through the investigation of their structure-activity relationship (SAR).
AUTHOR CONTRIBUTIONS
Conceptualization, Q.A.A. and S.M.A.; methodology, Q.A.A., S.M.A., and R.Y.B.; investigation, Q.A.A. and S.M.A.; data curation, Q.A.A. and R.Y.B.; formal analysis, Q.A.A. and R.Y.B; writing—original draft preparation, Q.A.A.; writing—review and editing, S.M.A. and R.Y.B. All the authors discussed the results and commented on the manuscript. All authors have read and agreed to the published version of the manuscript.
FINANCIAL SUPPORT
The authors wish to thank the Deanship of Scientific Research at Jordan University of Science and Technology for the financial support, project No.: 172-2023, and the Ministry of Higher Education, Scientific Research Fund, project No. MPH/1/14/2018.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. WHO. World Health Organization. Available from: https://www.who.int/news-room/fact-sheets/detail/cancer. Accessed on 28th July 2023.
2. Saini A, Kumar M, Bhatt S, Saini V, Malik, A. Cancer causes and treatments. Int J Pharm Sci Res. 2020;11:3121–34.
3. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18(1–2):43–73. CrossRef
4. Aziz NM, Rowland JH. Trends and advances in cancer survivorship research: challenge and opportunity. Semin Radiat Oncol. 2003 Jul;13(3):248–66. CrossRef
5. Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990 Jul 1;269(1):1–11. CrossRef
6. Thornalley PJ. The glyoxalase system in health and disease. Mol Aspects Med. 1993;14(4):287–371. CrossRef
7. Morgenstern J, Campos Campos M, Nawroth P, Fleming T. The glyoxalase system-new insights into an ancient metabolism. antioxidants (basel). 2020 Oct 1;9(10):939. doi: 10.3390/antiox9100939 CrossRef
8. Antognelli C, Talesa VN. Glyoxalases in urological malignancies. Int J Mol Sci. 2018 Jan 31;19(2):415. CrossRef
9. Chang T, Wu L. Methylglyoxal, oxidative stress, and hypertension. Can J Physiol Pharmacol. 2006 Dec;84(12):1229–38. CrossRef
10. Toyoda Y, Erkut C, Pan-Montojo F, Boland S, Stewart MP, Müller DJ, et al. Products of the Parkinson’s disease-related glyoxalase DJ-1, D-lactate and glycolate, support mitochondrial membrane potential and neuronal survival. Biol Open. 2014 Jul 25;3(8):777–84. CrossRef
11. Rabbani N, Thornalley PJ. Glyoxalase 1 modulation in obesity and diabetes. Antioxid Redox Signal. 2019 Jan 20;30(3):354–374. CrossRef
12. Rabbani N, Thornalley PJ. Glyoxalase in diabetes, obesity and related disorders. Semin Cell Dev Biol. 2011 May;22(3):309–17. CrossRef
13. Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs. 2008 Oct;17(10):1533–45. CrossRef
14. Geng X, Ma J, Zhang F, Xu C. Glyoxalase I in tumor cell proliferation and survival and as a potential target for anticancer therapy. Oncol Res Treat. 2014;37(10):570–4. CrossRef
15. Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996 Jun;27(4):565–73. CrossRef
16. Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules. 2012 Nov 22;17(12):13740–58. CrossRef
17. Al-Balas QA, Hassan MA, Al-Shar’i NA, Mhaidat NM, Almaaytah AM, Al-Mahasneh FM, et al. Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents. Drug Des Devel Ther. 2016 Aug 17;10:2623–9. CrossRef
18. Cameron AD, Olin B, Ridderström M, Mannervik B, Jones TA. Crystal structure of human glyoxalase I--evidence for gene duplication and 3D domain swapping. EMBO J. 1997 Jun 16;16(12):3386–95. CrossRef
19. Chiba T, Ohwada J, Sakamoto H, Kobayashi T, Fukami TA, Irie M, et al. Design and evaluation of azaindole-substituted N-hydroxypyridones as glyoxalase I inhibitors. Bioorg Med Chem Lett. 2012 Dec 15;22(24):7486–9. CrossRef
20. Yadav A, Kumar R, Sunkaria A, Singhal N, Kumar M, Sandhir R. Evaluation of potential flavonoid inhibitors of glyoxalase-I based on virtual screening and in vitro studies. J Biomol Struct Dyn. 2016 May;34(5):993–1007. CrossRef
21. Al-Balas QA, Hassan MA, Al-Shar’i NA, Al Jabal GA, Almaaytah AM. Recent advances in glyoxalase-I inhibition. Mini Rev Med Chem. 2019;19(4):281–91. CrossRef
22. Santarius T, Bignell GR, Greenman CD, Widaa S, Chen L, Mahoney CL, et al. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer. 2010 Aug;49(8):711–25. CrossRef
23. Bhat AA, Uppada S, Achkar IW, Hashem S, Yadav SK, Shanmugakonar M, et al. Tight junction proteins and signaling pathways in cancer and inflammation: a functional crosstalk. Front Physiol. 2019 Jan 23;9:1942. CrossRef
24. Yang YX, Chen ZC, Zhang GY, Yi H, Xiao ZQ. A subcelluar proteomic investigation into vincristine-resistant gastric cancer cell line. J Cell Biochem. 2008 Jun 1;104(3):1010–21. CrossRef
25. Audat SA, Al-Balas QA, Al-Oudat BA, Athamneh MJ, Bryant-Friedrich A. Design, synthesis and biological evaluation of 1,4-benzenesulfonamide derivatives as glyoxalase I inhibitors. Drug Des Devel Ther. 2022 Mar 28;16:873–85. CrossRef
26. Jin T, Zhao L, Wang HP, Huang ML, Yue Y, Lu C, Zheng ZB. Recent advances in the discovery and development of glyoxalase I inhibitors. Bioorg Med Chem. 2020 Feb 15;28(4):115243. CrossRef
27. Usami M, Ando K, Shibuya A, Takasawa R, Yokoyama H. Crystal structures of human glyoxalase I and its complex with TLSC702 reveal inhibitor binding mode and substrate preference. FEBS Lett. 2022 Jun;596(11):1458–67. CrossRef
28. Lavi A, Ngan CH, Movshovitz-Attias D, Bohnuud T, Yueh C, Beglov D, et al. Detection of peptide-binding sites on protein surfaces: the first step toward the modeling and targeting of peptide-mediated interactions. Proteins. 2013 Dec;81(12):2096–105. CrossRef
29. Bacilieri M, Moro S. Ligand-based drug design methodologies in drug discovery process: an overview. Curr Drug Discov Technol. 2006 Sep;3(3):155–65. CrossRef
30. Wilson GL, Lill MA. Integrating structure-based and ligand-based approaches for computational drug design. Future Med Chem. 2011 Apr;3(6):735–50. CrossRef
31. Kutlushina A, Khakimova A, Madzhidov T, Polishchuk P. Ligand-based pharmacophore modeling using novel 3D pharmacophore signatures. Molecules. 2018 Nov 27;23(12):3094. Erratum in: Molecules. 2019 Mar 18;24(6). CrossRef
32. Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today. 2010 Jun;15(11–12):444–50. CrossRef
33. Debnath AK. Pharmacophore mapping of a series of 2,4-diamino-5-deazapteridine inhibitors of Mycobacterium avium complex dihydrofolate reductase. J Med Chem. 2002 Jan 3;45(1):41–53. CrossRef
34. Youngstrom EA. A primer on receiver operating characteristic analysis and diagnostic efficiency statistics for pediatric psychology: we are ready to ROC. J Pediatr Psychol. 2014 Mar;39(2):204–21. CrossRef
35. Vyas VK, Ghate M, Goel A. Pharmacophore modeling, virtual screening, docking and in silico ADMET analysis of protein kinase B (PKB β) inhibitors. J Mol Graph Model. 2013 May;42:17–25. CrossRef
36. Zhou Y, Di B, Niu MM. Structure-based pharmacophore design and virtual screening for novel tubulin inhibitors with potential anticancer activity. Molecules. 2019 Sep 1;24(17):3181. CrossRef
37. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015 May;10(5):449–61. CrossRef
38. Tirado-Rives J, Jorgensen WL. Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J Med Chem. 2006 Oct 5;49(20):5880–4. CrossRef
39. Al-Balas QA, Al-Sha’er MA, Hassan MA, Al Zou’bi E. Identification of the first “two digit nano-molar” inhibitors of the human glyoxalase-I enzyme as potential anticancer agents. Med Chem. 2022;18(4):473–83. CrossRef