
INTRODUCTION
Diabetes is a chronic disease caused by either insufficient use of insulin by the body or insufficient production of insulin by the pancreas. The hormone insulin is known to control blood glucose levels. High blood sugar, commonly referred to as hyperglycemia, is a common side effect of uncontrolled diabetes. Many internal systems, including blood vessels and nerve cells, can be severely affected [1]. The severity of symptoms depends on the type and duration of diabetes. Particularly in children with complete insulin deficiency, other people with high blood glucose levels may develop polyuria, polydipsia, polyphagia, weight loss, and blurred vision [2]. Type 2 DM, the most common form of the disease, affects more than 90% of people [3] and their cells do not respond to insulin or produce insulin. The process typically starts with insulin resistance, a condition in which cells use insulin incorrectly. The pancreas gradually stops producing insulin as the body’s need for the hormone increases [4].
The main protein, glucose transporter 4 (GLUT4), has been identified as a therapeutic target for pharmaceutical intervention strategies to reduce hyperglycemia. GLUT4 can facilitate blood glucose uptake by muscle and adipose tissue cells [5]. According to research by Song et al. [6], Sophora alopecuroides L. produces quinolizidine alkaloid aloperine (ALO), which has been shown to warrant investigation as a potential hypoglycaemic agent. Protein kinase C (PKC) is activated by the G-protein-PLC-IP3R-Ca2+ signaling pathway, so this drug lowers blood glucose levels by inducing the plasma membrane of GLUT4 to fuse and absorb glucose.
An indigenous herb from Indonesia called Physalis angulata L, popularly known as Ciplukan, may have antiproliferative, diabetic, and anticytotoxic properties [7]. The herbs (all parts of the plant) of the Ciplukan plant are the most widely used constituents. Some people also use the roots, stems, fruits, and leaves of the plant. They contain chemicals from the terpenoid, alkaloid, phenolic, flavonoid, and saponin groups [8]. In contrast to the other fractions, fraction I of the P. angulata chloroform extract had the highest percentage of glucose reduction (26.47%), according to other previous work [9]. Due to the rapid decisions, in silico drug development is being evaluated worldwide. Bioinformatics and computational biology, combined with silico medicinal chemistry, are part of the current revolution in drug development. From the early stages of preclinical development to the later stages of clinical development, in silico approaches are essential. As well as speeding up the drug discovery process, it can also help to avoid costly late-stage clinical failures [10].
The problem arises from inadequate insulin production by the pancreas, resulting in decreased GLUT4 expression and subsequently reduced muscle glucose uptake. The aim of this study was to identify specific compounds by GC-MS analysis and to evaluate their potential as antihyperglycaemic agents in P. angulata, with a particular focus on their interaction with the target Akt substrate of 160 kDa (AS160). AS160, which contains a Rab (GTPase-activating protein (GAP) domain, is phosphorylated at multiple sites by the protein kinase Akt. In adipocytes, the insulin-triggered translocation of the glucose transporter GLUT4 to the cell membrane depends on the phosphorylation of AS160. This complex process of GLUT4 translocation and glucose absorption is tightly regulated by AS160 splicing [11,12]. The lack of mainstream diabetes drugs targeting AS160 is unfortunate. Our study took an unprecedented approach to this protein to assess its viability and potential applicability in future endeavors. The study by Kane et al. [13] showed that the molecular characteristics of AS160 are consistent with those of a Rab-GAPase activating protein (GAP) and underlined its central role as a regulator of GLUT4 traffic. AutoDock Vina (Vina), as described by Trott and Olson [14], is a commonly used method, especially when binding site details are available to ensure the effectiveness of the docking process. However, in scenarios where the specific protein binding pocket remains unidentified, Vina excels in performing blind docking (BD) procedures. BD involves enclosing the target in a unified simulation box, effectively covering the entire surface area of the protein [15]. The study aims to advance the search for potential lead compounds by predicting biological activity, gaining new perspectives on binding mechanisms, and anticipating pharmacological profiles.
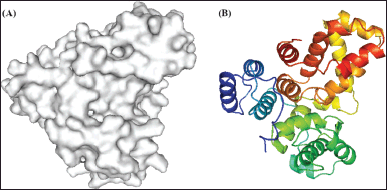 | Figure 1. Representation of 3QYB Chain A structure (A) surface; (B) Secondary structure. [Click here to view] |
MATERIALS AND METHODS
Active ingredient of P. Angulata L
The maceration method was used for extraction, in which 500 g of P. angulata powder was macerated with 2 l of chloroform. The compound was then obtained using vacuum liquid chromatography and preparative thin layer chromatography methods. Although the specific details of the bioactive compound are not included in this article, its antihyperglycaemic activity was evaluated using a glucose consumption assay. Appropriate measures were taken to prepare the active ingredient of P. angulata for gas chromatography-mass spectrometry (GC-MS) analysis.
GC-MS analysis and compound identification
A Thermo Scientific TM TRACE 1313 GC and a Thermo Scientific TM ISQ LT single quadrupole mass spectrometer were used for GC-MS analysis. The GC-MS system, operating in EI mode, has an HP 5MS UI capillary column with a 30 m × 0.25 mm × 0.25 m layer thickness. Helium UHP (He) was used as the carrier gas (1 ml minute 1). The temperature gradient was set to increase by 3°C per minute from 60°C to 280°C. The sample solution in a vial containing chloroform solvent was filtered and injected into a GC-MS instrument. The structure of the compound was identified by comparison with spectra from the Wiley 275 MS library and publicly available data. Molecular docking analysis was performed using the information from the GC-MS analysis.
Pre-processing of target structure
The 3D structure of human TBC1 domain family member 4 (TBC1D4) was obtained from the Protein Data Bank (https://www.rcsb.org; accessed on 28 July 2023), with a PDB ID of 3QYB (Fig. 1) and a resolution of 3.50 Å. The selection of this protein was based on its possession of the only available crystal structure known in the literature, specifically to accurately represent the AS160 receptor in docking studies [16]. Uniprot.org, under code O60343, confirmed that this structure corresponds to the only chain A structure known for TBC1D4 (accessed 28 July 2023). After visualization of the target structure and elimination of water molecules, the final molecule was saved in a (.pdb) file using PyMol. Autodock Tools 1.5.7 was then used to determine the grid box size, introduce charges, and predict the binding site using BD techniques. The resulting file was then converted into a molecular docking file (.pdbqt) [14].
 | Table 1. Isolated compounds from herbs of P. angulata L. [Click here to view] |
BD to predict the binding site
The ligand, derived from natural products and purchased from PubChem (CID 5546) [17,18], underwent 2D structural optimization facilitated by Open Babel [19]. Adjustments were made to the protein lattice box size to match the chain dimensions. A 3QYB structure was constructed using Autodock Tools 1.5.7 [14]. Following ligand optimization, which resulted in (.pdbqt) file extensions, the docking process involved the generation of 20 modes and redocking twice, resulting in a final production of 60 modes using Autodock Vina. Visualization of the docking results was performed using Pymol [18], allowing identification of the prominent binding site favored by the ligand. This identified binding site will be used in the molecular docking analysis of potential compounds from P. angulata.
Molecular docking of compounds against AS160 protein
The active compound identified by bioassay-guided isolation (see Table 1) was successfully isolated in a previous study (not presented here) and characterized by GC-MS analysis. All ligands from the PubChem database (accessed on 30 July 2023) were subjected to energy minimization using Open Babel. The ligand formats were converted from .sdf to (.pdbqt) and AutoDock Vina was used to generate the 3QYB target structure. Through cluster analysis within the protein target structure, the docking positions of the ligands were visualized using PyMol and LigPlot+. The resulting complexes were then used to elucidate the binding process.
Binding characterisation mechanism
LigPlot+ (v.2.2.8) was used to look at the hydrogen bonding and hydrophobic interactions between the selected drugs and the target protein structure. Interactions between the selected compounds and known inhibitors with the target structure were examined to find common residues that may be critical for ligand binding.
RESULTS AND DISCUSSION
Target structure pre-processing
The 3D structure of the AS160 protein, also known as Human TBC1D4 PDB ID: 3QYB, was obtained. The structure was selected as the best structure from the UniProt list with the highest resolution, 3.50 Å. The AS160 gene sequence from The Research Collaboratory for Structural Bioinformatics is still bound to water molecules and other solvents and is eliminated. AS160, a protein that activates Rab GTPases, appears to be phosphorylated and inactivated by Akt [16]. This protein activates a GTP-bound form of an unnamed Rab protein. It may also regulate one or more steps in the translocation of GLUT4 vesicles to the cell surface (Fig. 2).
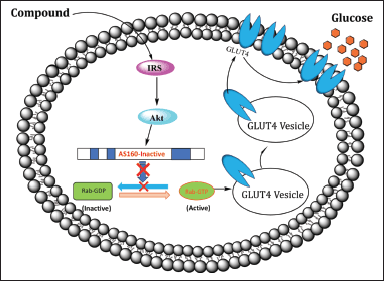 | Figure 2. Schematic of the GLUT4 translocation to the cell surface of the insulin-signaling cascade. [Click here to view] |
 | Table 2. Ligand for BD. [Click here to view] |
BD to predict the binding site
Due to the lack of research on the AS160 protein structure in the UniProt database, coupled with the lack of information on the target pocket or binding site in the 3D structure, molecular docking was performed using the BD method [20]. The use of BD expands the conformational search space and improves docking accuracy by not restricting binding to a specific protein pocket. For binding site prediction, a grid size was created that included all 3QYB proteins with coordinates that covered the protein surface (X: −57.781; Y: 55.459; Z: 6.341). This coordination center was determined after prediction to identify unknown binding pockets and minimize the search space. Ligands derived from natural sources were used to randomly bind to the targets, with triamterene (see Table 2) being selected. Triamterene was chosen randomly as the ability of native ligands to interact with the AS160 protein remains unknown. In addition, numerous studies have investigated the potential effects of triamterene [21,22].
Molecular docking was performed using Autodock Vina with 20 output modes and a completeness of 64. Two subsequent redockings, each with 60 output ligands, were used to identify the potential binding sites on the protein. The ligand structure showed a distribution over 8 sites, but two primary centers with the highest probability were identified. These are binding site 1 (X: −65.295; Y: 50.280; Z: 24.497), which represents 36.6%, and binding site 2 (X: −49.276; Y: 60.786; Z: −1.742), which represents 26.6% (see Fig. 3). The remaining 36.8% of the ligand structures were distributed over 6 other sites not presented in this paper. The determined binding site dimensions are subsequently used to perform docking analyses between the AS160 protein and potential compounds derived from P. angulata.
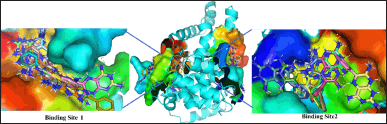 | Figure 3. The binding site included in the blind-docking simulation. [Click here to view] |
Molecular docking of compounds against the AS160 protein
The molecular docking ligands were obtained from P. angulata L. herbs by GC-MS analysis, as shown in Table 3, and their molecular weight was validated by mass spectra (MS). Notably, among the different fractions, fraction I of the chloroform extract of P. angulata showed the highest percentage of glucose reduction (26.47%) [9]. Our previous unpublished research confirmed the successful isolation of bioactive compounds from fraction 1 using a bioassay-guided isolation strategy. GC-MS analysis revealed that the isolated compound from P. angulata consists of a mixture of known phytosterols, campesterol, and stigmasterol (see Table 3). Stigmasterol has a retention time (RT) of 31.889, whereas the RT of campesterol is 31.298. With stigmasterol having an m/z of 412 and campesterol having an m/z of 400, both compounds have similarity index match values of over 90% (see Fig. 4). Therefore, this study aims to investigate the in silico antidiabetic properties of stigmasterol and campesterol.
The glucose transporter GLUT4 is present in various tissues, including skeletal muscle, heart, adipose tissue, and brain, and is responsive to insulin. GLUT4 is located in the cytoplasmic vesicles of cells and its translocation to the cell membrane is initiated by insulin binding to insulin receptors [23]. Insulin resistance, associated with diabetes mellitus (DM), refers to a diminished response to insulin. In response to this inefficiency, the body increases insulin production in an attempt to restore glucose homeostasis. Type 2 DM (T2DM) is a consequence of this increased insulin production, which subsequently declines. Numerous studies have explored targeting the glucose transporter GLUT4 as a therapeutic approach to T2DM, with the aim of increasing GLUT4 protein translocation and expression [23]. Potential pathways that have been investigated include the PI3K/Akt pathway and the G protein-PLC-PKC pathway through the target protein [6,11]. Our study focused on phytosterol compounds extracted from P. angulata and investigated their potential to alleviate hyperglycemia through in silico experiments. Consequently, we performed in silico experiments to elucidate the effects of campesterol and stigmasterol on T2DM.
 | Table 3. Chemical profiling of bioactive compounds in P. angulata L by using GC-MS analysis. [Click here to view] |
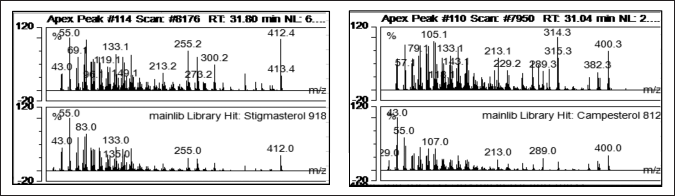 | Figure 4. Mass fragmentograms (m/z 412) of the characteristic ion for (A) stigmasterol and mass fragmentograms (m/z 400) of the characteristic ion for (B) campesterol by GC-MS analysis. [Click here to view] |
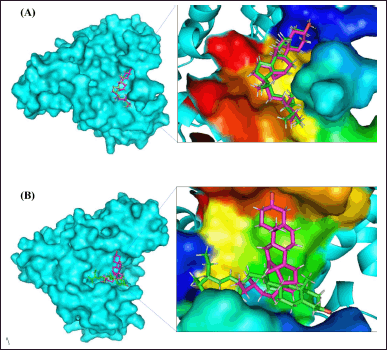 | Figure 5. A visual representation of the compounds found in P. angulata L and their docking to the active site of AS160 would provide an insightful schematic. This scheme could show the molecular interactions and positioning of these compounds within the active site of the AS160 receptor, elucidating their potential binding and functional relationships. (A) Binding site 1. (B) Binding site 2 (note: magenta as campesterol and green as stigmasterol). [Click here to view] |
The major glucose transporter in skeletal muscle, known as GLUT4, is central to understanding the pathophysiology of molecular docking. To comprehensively cover the active site residues, a single grid box was used, derived from a BD investigation, taking into account potential secondary substrate binding sites. PyMol proved to be a valuable tool for investigating protein-ligand interactions and assessing the strength of ligand binding within the active region. As shown in Figure 5, almost all of the chemicals were docked deep within the binding site of the AS160 receptor. Cluster analysis of the docking results played a crucial role in identifying the optimal poses for each molecule. This analytical approach provides insight into the most likely binding sites for protein-ligand combinations [24].
LigPlot+ was used to visualize the protein-ligand interactions. The study of molecular interactions is crucial for understanding biological regulatory mechanisms and providing a theoretical basis for the development of new therapeutic targets [25]. Both strong hydrophobic contacts and delicate intermolecular interactions, such as hydrogen bonding, play a role in stabilizing ligands at active sites and can serve as indicators of binding affinity and efficacy [26]. Conditions indicative of hydrogen bonding in interaction studies require a hydrogen donor and acceptor with a bond distance of less than 3.9 Å, while angles greater than 150° indicate the strength of the hydrogen bond [27,28]. The following table provides details on hydrogen bonding and hydrophobic interactions for all three compounds in complex with the AS160 receptor at binding sites 1 and 2 (see Table 5).
In molecular docking studies at binding site 1, both campesterol and stigmasterol showed hydrogen bond interactions with the protein target. Specifically, both compounds formed two hydrogen bonds with the active site residues His940 and Arg941. The bond lengths for campesterol were measured to be 3.12 and 3.03 Å, whereas stigmasterol exhibited shorter bond lengths of 3.02 and 2.80 Å (see Fig. 6A and B, respectively). Given the preference for shorter bond lengths, as they improve subsequent binding, it could be argued that stigmasterol is favored over campesterol.
 | Table 4. ADME properties of phytochemical compounds predicted by SwissADME. [Click here to view] |
 | Table 5. Binding energies of compounds and proteins with their corresponding hydrogen and hydrophobic interacting residues. [Click here to view] |
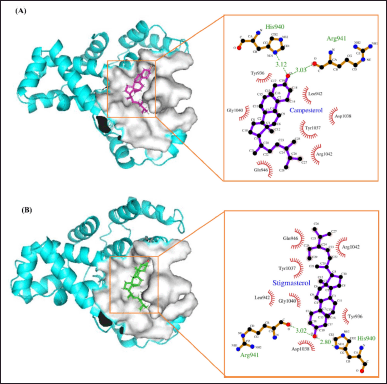 | Figure 6. Schematic and Ligplot+ representation of the interaction mechanism of (A) campesterol and (B) stigmasterol in the active site of AS160 at binding site 1. [Click here to view] |
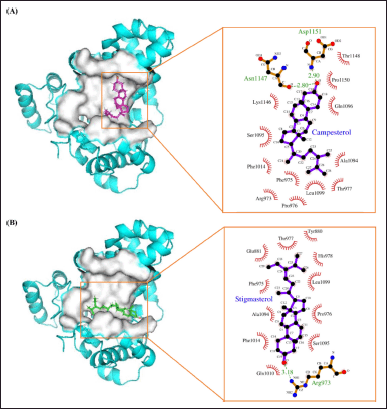 | Figure 7. Schematic and Ligplot+ representation of the interaction mechanism of (a) campesterol and (b) stigmasterol in the active site of AS160 at binding site 2. [Click here to view] |
In addition, hydrogen bonds were formed at binding site 2, similar to the previous site, involving protein amino acids. Campesterol, among other ligands, exhibited the shortest bond lengths of 2.80 and 2.90 Å, respectively. It formed two hydrogen bonds with the active site residues Asn1147 and Asp1151 (see Fig. 7A). In contrast, the other ligands exhibited longer bond lengths above 3 Å. Stigmasterol formed a hydrogen bond with Arg973 at a length of 3.18 Å (see Fig. 7B). Importantly, all bond lengths remained within an acceptable threshold. In conclusion, the interaction of the two compounds at binding site 2 suggests a high level of selectivity for these molecules [29]. The main binding interactions between the compounds and the active site residues are primarily facilitated by hydrogen bonds, which ensure the stability of the complexes [30].
Based on the results of a molecular docking simulation, the phytosterols found in Ciplukan (P. angulata) show potential in the treatment of type 2 diabetes by targeting the glucose transporter GLUT4 through the process of GLUT4 translocation. These compounds hold promise as a reliable source of bioactive compounds for antihyperglycaemic agents, offering a potential avenue for the prevention of DM. This finding is consistent with previous research where stigmasterol isolated from the seaweed Gelidium spinosum showed reduced hyperglycaemic effects, as confirmed by in vitro and in vivo studies [31]. Furthermore, edible soy oil containing stigmasterol significantly increased GLUT4 translocation and glucose uptake in L6 cells and skeletal muscle. In addition, oral stigmasterol therapy in KK-Ay rats improved insulin resistance and oral glucose tolerance as evidenced by reduced fasting blood glucose levels and improved blood lipid indicators such as triglycerides and cholesterol [32].
Plant-derived substances such as campesterol, stigmasterol, and β-sitosterol which have structural similarities to cholesterol, play crucial roles in structural stability, regulatory functions, and signaling [33]. Building on the above explanation, molecular docking, a simulation technique that focuses on the interaction between two molecules, provides insight into the potential efficacy of a potential drug. However, experimental demonstrations in the laboratory are essential to validate the results of in silico studies, especially with regard to pharmacological effects such as in vivo responses and toxicity.
We predicted absorption, distribution, metabolism, and excretion (ADME) properties for a more holistic simulation using SwissADME prediction. Campesterol and stigmasterol behave as drug-like, moderately soluble in water, topological polar surface area (TPSA) (20.23 Å2) and 1 violation of Lipinski’s rule of five, making them potential candidates as antihyperglycaemic agents (Table 4). We also advocate the use of molecular dynamics (MDs) simulations to investigate the stability of complex structural interactions. This approach facilitates the assessment of conformational stability, residual flexibility of amino acids, and consistency of hydrogen bonding profiles over the simulation period. As reported by Wahyudi et al. [34], natural compounds (cosmosiin, glucobrassicin, and isobavachin) exhibited MDs simulations and ADME properties comparable to those of recognized Mpro inhibitors of SARS-CoV-2, providing a comprehensive simulation.
CONCLUSION
The isolated compounds from P. angulata L herbs, namely campesterol and stigmasterol, show potential inhibitory activity with the AS160 protein, which is thought to play a role in regulating GLUT4 vesicle translocation to the cell surface. These phytoconstituents were found to have promising pharmacokinetic properties, making them an excellent source of naturally occurring chemicals with hypoglycaemic activity. However, further in vivo investigations are essential to validate these findings and assess their potential utility in the management of DM.
ACKNOWLEDGMENTS
The authors would like to thank Nurani Alawiyah for her technical assistance. This work was funded by the project Hibah Dana Masyarakat (DAMAS) Project, Faculty of Medicine, Public Health and Nursing, Gadjah Mada University, 2023.
AUTHOR CONTRIBUTIONS
Rita Rakhmawati: Concept and design, data collection, data analysis, drafting of the manuscript, and final approval. Setyanto Tri Wahyudi: Concept and design, data collection, data analysis, drafting of the manuscript, critical revision of the manuscript, supervision, and final approval. Mae Sri Hartati Wahyuningsih: Concept and design, data collection, drafting of the manuscript, critical revision of the manuscript, funding, supervision, and final approval. Mustofa: Concept and design, collection, critical revision of the manuscript, supervision, and final approval. Ahmad Hamim Sadewa: Concept and design, data collection, critical revision of the manuscript, supervision, and final approval.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. World Health Organization. Diabetes [Internet]. Geneva: WHO; 2023 [cited 2023 July 25]. Available from: https://www.who.int/news-room/fact-sheets/detail/diabetes
2. Kharroubi AT, Hisham MD. Diabetes mellitus: the epidemic of the century. World J Diabetes. 2015;6(6):850–67. CrossRef
3. Centers for Disease Control and Prevention. What is Diabetes? [Internet]. Atlanta: CDC. 2023 [cited 2023 May 19] Available from: https://www.cdc.gov/diabetes/basics/diabetes.html#:~:text=With%20diabetes%2C%20your%20body%20doesn,vision%20loss%2C%20and%20kidney%20disease
4. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):1–34. CrossRef
5. Carvalho E, Kotani K, Peroni OD, Kahn BB. Adipose-specific overexpression of GLUT4 reverses insulin resistance and diabetes in mice lacking GLUT4 selectively in muscle. J Physiol Endocrinol Metab. 2005;289(4): 551–61. CrossRef
6. Song G, Huang Y, Xiong M, Yang Z, Liu Q, Shen J, et al. Aloperine relieves type 2 diabetes mellitus by enhancing GLUT4 expression and translocation. Front Pharmacol. 2021;11:1–15. CrossRef
7. Saraswati E, Wijaya AS. Antibacterial activities of Physalis angulata herb extract on white feces diseases (WFD) in Litopenaeus shrimp vannamei. IOP Conf. Ser.: Earth Environ. Sci. 2019;236(1):0–6. CrossRef
8. Panjaitan RGP, Titin T, Yuliana YGS. Description of ciplukan toxicity (Physalis angulata L.). Pharmacogn J. 2023;15(3):357–67. CrossRef
9. Wahyuningsih MSH, Ketut SSW, Aurelia PRP, Nugrahaningsih DAA, Yuniyanti MM. Bioassay-guided fractionation of ciplukan (Physalis angulata L.) monitored by glucose consumption assay and thin layer chromatography on myoblast cells. Majalah Obat Tradisional. 2023;28(1):22–30. CrossRef
10. Lisdiana L, Widiatningrum T, Kurniawati F. Molecular docking bioactive compound of rambutan peel (Nephelium lappaceum L) and NF-Κb in the context of cigarette smoke-induced inflammation. TJNPR. 2022;6(10):1654–59. CrossRef
11. Baus D, Heermeier K, De-Hoop M, Metz-Weidmann C, Gassenhuber J, Dittrich W, et al. Identification of a novel AS160 splice variant that regulates GLUT4 translocation and glucose uptake in rat muscle cells. Cell Signal. 2008;20(12):2237–46. CrossRef
12. Mîinea CP, Sano H, Kane S, Sano E, Fukuda M, Peränen J, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391(1):87–93. CrossRef
13. Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277(25):22115–8. CrossRef
14. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31(2):455–61. CrossRef
15. Muscat S, Pallante L, Stojceski F, Danani A, Grasso G, Deriu MA. The impact of natural compounds on S-shaped Aβ42 fibril: from molecular docking to biophysical characterization. Int J Mole Sci. 2017;21(6):1–15. CrossRef
16. Park SY, Jin W, Woo JR, Shoelson SE. Crystal structures of human TBC1D1 and TBC1D4 (AS160) RabGTPase-activating protein (RabGAP) domains reveal critical elements for GLUT4 translocation. J Biol Chem. 2011;286(20):18130–38. CrossRef
17. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–109. CrossRef
18. Seeliger D, De Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–22. CrossRef
19. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3(33):1–14. Available from: https://jcheminf.biomedcentral.com/track/pdf/10.1186/1758-2946-3-33
20. Zhang W, Bell EW, Yin M, Zhang Y. EDock: blind protein-ligand docking by replica-exchange Monte Carlo simulation. J Cheminform. 2020;12(1):1–17. CrossRef
21. Li Y, Cheng KC, Niu CS, Lo SH, Cheng JT, Niu HS, et al. Investigation of triamterene as an inhibitor of the TGR5 receptor: identification in cells and animals. Drug Des Devel Ther. 2017;11:1127–34. CrossRef
22. Niyazov R, Sharman T. Triamterene. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 May 22]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557650/
23. Navale A. Glucose transporters and their cellular form, role and function. In Molecular Nutrition Carbohydrates. Amsterdam: Elsevier Inc. 2019. CrossRef
24. Taha M, Shah SAA, Afifi M, Imran S, Sultan S, Rahim F, et al. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg Chem. 2018;77:586–92. CrossRef
25. Fu Y, Zhao J, Chen Z. Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: a case of oligopeptide binding protein. Comput Math Methods Med. 2018. CrossRef
26. Varma AK, Patil R, Das S, Stanley A, Yadav L, Sudhakar A, et al. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS One. 2010;5(8). CrossRef
27. Bartlett GJ, Newberry RW, Vanveller B, Raines RT, Woolfson DN. Interplay of hydrogen bonds and n → π* interactions in proteins. J Am Chem Soc. 2013;135(49):18682–88. CrossRef
28. Menéndez CA, Accordino SR, Gerbino DC, Appignanesi GA. Hydrogen bond dynamic propensity studies for protein binding and drug design. PLoS One. 2016;11(10):1–12. CrossRef
29. Aldahish A, Balaji P, Vasudevan R, Kandasamy G, James JP, Prabahar K, et al. Elucidating the potential inhibitor against type 2 diabetes mellitus associated gene of GLUT4. J Pers Med. 2023;13(4). CrossRef
30. Adinortey CA, Kwarko GB, Koranteng R, Boison D, Obuaba I, Wilson MD, et al. Molecular structure-based screening of the constituents of Calotropis procera identifies potential inhibitors of diabetes mellitus target alpha-glucosidase. Curr Issues Mol Biol. 2022;44(2):963–87. CrossRef
31. Poulose N, Sajayan A, Ravindran A, Chandran A, Priyadharshini GB, Selvin J, et al. The anti-diabetic potential of stigmasterol from the seaweed Gelidium spinosum and its application in the formulation of nanoemulsion conjugate for the development of functional biscuits. Front Nutr. 2021;1–10. CrossRef
32. Muñoz-Gómez RJ, Rivero-Cruz I, Ovalle-Magallanes B, Linares E, Bye R, Tovar AR, et al. Antidiabetic Sterols from Peniocereus greggii Roots. ACS Omega. 2022;7(15):13144–54. CrossRef
33. Jayaraman S, Roy A, Vengadassalapathy S, Sekar R, Veeraraghavan V, RajagopalP, et al. An overview of the therapeutic function of foods enriched with plant sterols in diabetes management. Antioxidants. 2021;10(12). CrossRef
34. Wahyudi ST, Muscifa ZS, Sumaryada T, Ambarsari L. Computational simulation of indonesian natural compounds as main protease. IJCB. 2023;2(1):11–21. CrossRef