
INTRODUCTION
Chloroacetyl chloride, often abbreviated as chloroacetyl chloride (CAC), is a well-established raw material essential in both the pharmaceutical and chemical industries. Its primary purpose lies in the synthesis of critical intermediates and active pharmaceutical ingredients. This compound, known for its highly reactive and corrosive nature, poses significant hazards when it comes into contact with the skin, leading to severe chemical burns. In addition, chloroacetyl chloride can readily permeate the skin and affect the respiratory system [1]. Notably, chloroacetyl chloride exhibits favorable solubility in both water and methanol [2,3]. Its molecular structure [4]. includes a reactive “acyl-chloride” functional group (COCl), rendering it highly reactive toward nucleophiles. This characteristic makes it particularly suitable for acylation reactions [5], wherein it reacts with nucleophiles to generate acyl derivatives. However, it is crucial to acknowledge its extreme toxicity and corrosiveness, as the U.S. Environmental Protection Agency classifies it as an exceedingly hazardous substance. Comparatively, MCA, another chemical compound is considered “toxic” but may possess a lower degree of hazard when compared to chloroacetyl chloride. MCA (CH3O2CCl) contains an ester functional group, which exhibits reduced reactivity in comparison to acyl chlorides. Esters can undergo nucleophilic substitution reactions, but their reactivity is generally lower due to the presence of an electron-withdrawing chlorine atom. Interestingly, there is a paucity of information regarding the genotoxicity of chloroacetyl chloride. Hence, we base the risk assessment on the principles of the threshold of toxicological concern [6–9] and the structure-activity relationship [10–20]. Chloroacetyl chloride is categorized as a class 3 substance. Considering its potential to react with DNA and its potential genotoxicity [21–23]. An evaluation in accordance with the International Conference of Harmonization (ICH) M7 guidelines is imperative [24–27]. Several analytical methods have been employed to analyze chloroacetyl chloride. For instance, Khan et al. [28] utilized a capillary zone electrophoretic method, Morissette et al. [29] employed a derivatization GC method with a limit of quantification (LOQ) of 0.03%w/w. Langvardt et al. [30] established a derivatization procedure using electron capture gas chromatography (GC). Ajit Anerao et al. published a GC technique for measuring the levels of chloroacetyl chloride and thionyl chloride in tadalafil [31]. Kennedy [32] studied quantitative analysis of acid chlorides using an automatic cold on-column injection method. Klein et al. [33] developed an high performance liquid chromatography (HPLC) method for determining chloroacetyl chloride in the air. McCullough et al. [34] documented their study involving the collection of chloroacetyl chloride from the air using solid support, with its quantification performed via ion chromatography. Zhou et al. [35] developed a chemical derivatization HPLC method for the determination of chloroacetyl chloride and chloroacetic acid in raw material of Azintamide. Furthermore, Langhorst [36] developed a resin-coated solid sorbent tube for monitoring air bon reactive chemicals including chloroacetyl chloride, acetic anhydride, and isocyanatoethyl methacrylate, derivatized using a reagent l-(2-pyridyl) piperazine, and analyzed using an high-performance thin layer chromatography chromatographic technique. To the best of my knowledge, researchers have not reported any methods for determining trace levels of chloroacetyl chloride in chlordiazepoxide hydrochloride.
A notable feature of chloroacetyl chloride, when used as a pharmaceutical raw material, is that it often remains in trace amounts in the final Active Pharmaceutical Ingredients, in the present study Figure 1 shows the structures of chloroacetyl chloride, MCA, and chlordiazepoxide hydrochloride
(APIs). This retention occurs due to the incomplete consumption of reagents in chemical reactions (as illustrated in Figure 1. In light of this, Liu et al. [37] emphasize the importance of sensitive analytical techniques for pharmaceutical genotoxic impurities (GTIs), especially when dealing with trace levels at ppm (parts per million) concentrations. The identification, determination, and management of GTIs in pharmaceutical substances have become a significant concern, particularly in the context of their potential association with human cancer [38–42]. In summary, derivatization procedures are widely used for CAC estimation, which enhances the sensitivity and selectivity of analytical techniques but comes with several drawbacks. They involve additional steps, leading to increased sample handling and the potential for errors and contamination. The derivatization process is time-consuming, adding to the overall analysis time and reducing sample throughput. Some analytes may be sensitive to the reagents and conditions, leading to degradation or decomposition [43,44]. Derivatization can increase the cost of analysis due to the need for specialized reagents and equipment, and it requires a high level of skill and expertise. Contamination risks are higher, and chemical waste is generated, adding to environmental concerns. Interference from the reagents and limitations on the applicability of derivatization to all compounds further compound these drawbacks, making it important to carefully consider its utility in specific analytical applications also this literature review underscores the need for rigorous assessment and control of traceable reagents in pharmaceutical production. These impurities, including chloroacetyl chloride, may pose unintended risks such as genotoxicity or carcinogenicity. Consequently, their concentrations must be minimized to levels deemed negligible in terms of human safety, even when complete elimination is unattainable. Chloroacetyl chloride, due to its high reactivity, can be converted to a more stable form, MCA, through a nucleophilic acylation reaction with methanol, as depicted in Figure 2. This transformation enables the subsequent determination of MCA using GC with a flame ionization detector method, offering a straightforward and sensitive means of indirectly estimating chloroacetyl chloride. This process has been verified through analytical techniques such as mass spectrometry, and gas chromatographic-flame ionization detection (GC-FID), with comparisons to standard MCA and CAC compounds.
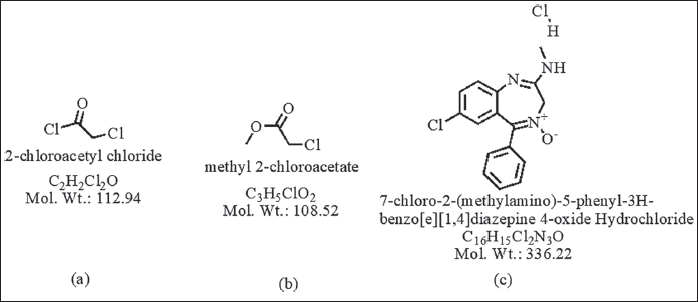 | Figure 1. The structure of (a) chloroacetyl chloride, (b) MCA, and (c) chlordiazepoxide hydrochloride. [Click here to view] |
In this study, chloroacetyl chloride underwent complete conversion to MCA in the presence of methanol, as demonstrated by GC. Notably, Figures 8 and 9 display matching retention times and peak areas for MCA and chloroacetyl chloride standards 1.5 ppm spiked in chlordiazepoxide hydrochloride, providing clear evidence of the successful transformation and validating the complete conversion. In addition, liquid chromatography-mass spectrometry (LC-MS) analysis reinforced these findings, with the mass spectrum aligning with the expected profile of MCA In the presence of Dichloromethane and Ethylene Dichloride, chloroacetyl chloride exhibits stability, as depicted in Figures 13 and 14. However, when subjected to methanol, it undergoes conversion to MCA, as evidenced by the LC-MS mass spectrum provided in the supplementary material. These results collectively offer robust scientific evidence for the efficient conversion of chloroacetyl chloride to MCA in the specified experimental conditions.
EXPERIMENTAL STUDY
Materials and methods
Chemicals and standards
We procured HPLC-grade methanol (≥99%) from standard reagents. Loba Chemie India supplied AR-grade chloroacetyl chloride (≥99%). We used MCA (98%), manufactured by AVRA. Flowchem Pvt. Ltd. generously provided chlordiazepoxide hydrochloride API-free samples, with the purity of the APIs exceeding 99.5%.
Instrumentation
We used a Shimadzu GC model The GC-2010 pro instrument featured a flame ionization detector and AOC20i is the auto-sampler model. The GC capillary column, DB Wax, with dimensions of 15 m length, 0.530 mm diameter, and a 1.0 µm film thickness, was sourced from Agilent Technologies. We employed a Shimadzu make weighing balance model AP225WD, with 0.1 mg accuracy. We used standard volumetric flasks and pipettes for standard and sample preparations.
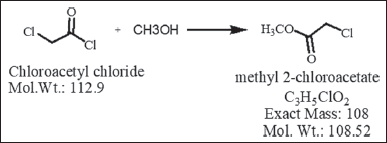 | Figure 2. Nucleophilic acylation reaction, chloroacetyl chloride reacts with methanol to produce MCA. [Click here to view] |
Chromatographic conditions
The final chromatographic conditions for the optimized method were established as follows: We utilized a DB Wax column with dimensions of 15 m in length and 0.53 mm in diameter, featuring a particle size of 1.0 µm. Initially, the oven temperature was set at 40°C for 5 minutes. Subsequently, it was ramped up at a rate of 10°C per minute until reaching 200°C, where it was maintained for 5 minutes. The injector temperature was set to 150°C, and the detector temperature was maintained at 230°C. The column flow rate was kept at 5 ml/minutes with a 2:1 split ratio. A 2 µl injection volume was introduced, using methanol as the diluent.
Preparing standard and sample solutions
To prepare the standard stock solution, we precisely weighed around 10 mg of “chloroacetyl chloride” and added it to a 100 ml standard volumetric flask. Then, we dissolved and diluted it with methanol until it reached the 100 ml mark, resulting in a solution with a concentration of 100 µg/ml for chloroacetyl chloride in the form of MCA.
To prepare the standard solution, we precisely transferred 1mL of the standard stock solution into a 100 ml standard flask and subsequently diluted it to the specified volume with methanol. A subsequent dilution of 0.75 ml of this solution with methanol in a 20 ml volumetric flask yielded a concentration of 37.5 ng/ml.
For the preparation of the test sample, we transferred 50 mg of chlordiazepoxide hydrochloride sample, dissolving it in 2 ml of methanol, thereby achieving a concentration of 25 mg/ml.
During the system suitability testing, we injected the 1.5 ppm standard solution six times in replicates, to ensure that the relative standard deviation (RSD) for the area response of the standard solution remained below 10.0%.
The LOD solution was prepared based on a signal-to-noise (S/N) ratio, yielding an observed lowest detectable concentration of 0.19 ppm, with an S/N ratio of 3.35.
To determine the LOQ solution, we applied a S/N ratio of 10.07, yielding the lowest quantifiable concentration of 0.38 ppm.
To assess linearity, we generated a series of solutions utilizing the standard stock solution, spanning a concentration range from 12.5% to 120%.
Method development
We aimed to quantify chloroacetyl chloride as Methyl 2-chloro acetate in “chlordiazepoxide hydrochloride” active drug substances during method development. Initially, the research was followed by a flow chart as mentioned in Figure 3, we made several attempts using different GC stationary phases, including DB-5, DB-624, and Rtx-225. Ultimately, the DB-Wax GC column yielded a suitable response from the analyte. We evaluated various diluents, such as methylene dichloride, ethylene dichloride, ethanol, dimethyl formamide, dimethyl Acetamide, and dimethyl sulfoxide. Methanol, chosen for its ability to convert chloroacetyl chloride to MCA, was the selected solvent. Initially, we used the GC column DB-5 containing 5%-phenyl-methyl polysiloxane stationary phase, and prepared a 1.5-ppm standard solution for injection into the chromatographic system. Although we detected 1.5 ppm of chloroacetyl chloride, the peak shape was asymmetric, and the area response was low. Subsequently, we tried the GC column DB-624 a mid-polar column containing “6% cyanopropyl/phenyl, and 94% polydimethylsiloxane” stationary phase, and again, 1.5 ppm of chloroacetyl chloride was detected, but with a low area response and an improper baseline. We then attempted the Rtx-225 column containing a “cross-bonded 50% cyanopropyl methyl/50% phenyl methyl” stationary phase, and similar challenges were encountered with a low area response and an improper baseline. Finally, we selected the GC column DB-Wax column containing “polyethylene glycol” stationary phase, and used methanol as the diluent to convert chloroacetyl chloride to MCA, resulting in a well-detected 1.5 ppm of MCA in methanol using the DB Wax GC column. The method development was further validated with the DB Wax capillary column and methanol as the diluent, providing superior peak shape and baseline separation. The retention time of MCA was observed at approximately 10.80 minutes.
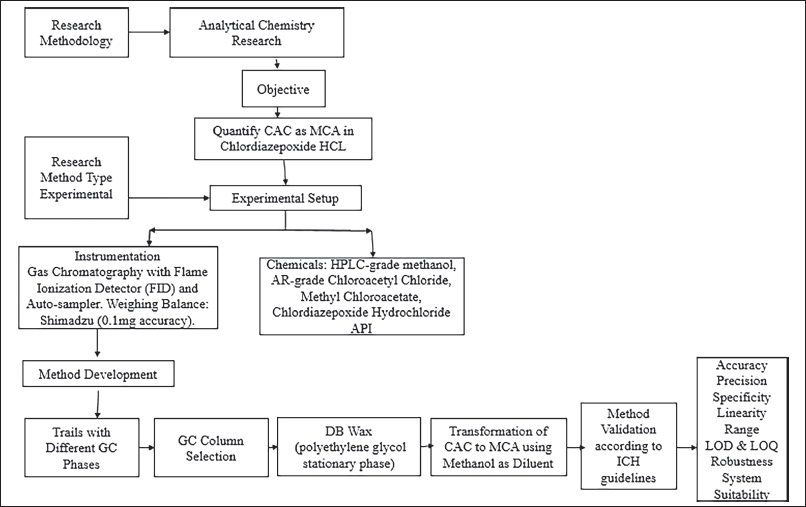 | Figure 3. Flow chart of experimental work. [Click here to view] |
RESULTS AND DISCUSSIONS
Analytical method validation
We conducted the validation process, following the ICH-Q2 (R1) guidelines [45]. To affirm the established GC method’s dependability for the indirect determination of chloroacetyl chloride content in chlordiazepoxide active drug compounds. Table 1 displays the percent RSD (percentage RSD) of MCA (1.5 ppm) from six replicates, demonstrating a percentage RSD of 1.386% when utilizing the DB Wax GC column. The validation, in alignment with ICH recommendations, encompassed the assessment of the following parameters: system suitability and specificity, accuracy, limits of quantification and detection, range and linearity, precision, and solution stability.
Specificity and system suitability
In the presence of potential interfering chemicals, specificity ensures precise measurement and differentiation of the target analyte, while system suitability validates the consistent and dependable performance of the analytical system. To confirm the proper functioning of the system and data generation, we prepared a 1.5-ppm methyl 2-chloro acetate standard solution in methanol and introduced it into the system six times. The percentage RSD for the six replicates of the standard solution was 1.386, significantly below the maximum acceptable threshold of 15.0%, thus confirming the system’s suitability.
In the assessment of method specificity, we also introduced various listed solvents; however, no interference was observed at the retention time of the analyte peak. Further testing revealed the absence of interference from the blank at the 10.8-minute retention time, corresponding to the methyl 2-chloro acetate standard peak. Consequently, we concluded that the MCA peak remained unaffected by the presence of diluent (blank) peaks.
Precision
The method’s precision demonstrates its ability to consistently generate reproducible and consistent results when analyzing the same sample under uniform conditions, indicating the degree of random error in measurements. In the system suitability results presented in Table 1, an RSD of 1.386 was obtained from six replicates of 1.5-ppm standard chloroacetyl chloride solution, demonstrating the accuracy of the method, to further assess precision; we prepared six homogeneous samples of chlordiazepoxide hydrochloride, calculating the percentage RSD for the content of chloroacetyl chloride in these samples. The chloroacetyl chloride content was found to be below the detection limit initially. Subsequently, we spiked known impurity chloroacetyl chloride up to the specification limit of 1.5 ppm to establish method precision. The percentage RSD for the content of chloroacetyl chloride in six preparations of spiked test samples was observed as 0.53%, which did not exceed the limit of 15.0%, as indicated in Table 2. Consequently, we have confirmed the method’s precision.
Detection and quantitation limits
We established the detection and quantitation limits through precise quantification of substances at the lowest levels within sample matrices. Utilizing the S/N ratio approach as presented in Table 3, we determined the limit of quantitation to be 0.38 ppm and the limit of detection to be 0.19 ppm and corresponding chromatograms as shown in Figures 4–6.
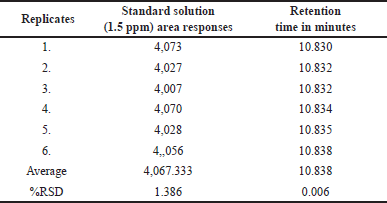 | Table 1. The precision table contains results from six replicates of the chloroacetyl chloride standard solution (1.5 ppm). [Click here to view] |
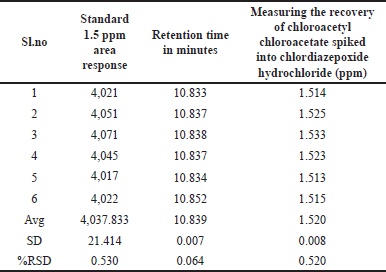 | Table 2. Spiked chloroacetyl chloride into chlordiazepoxide hydrochloride at a specification level of 1.5 ppm. [Click here to view] |
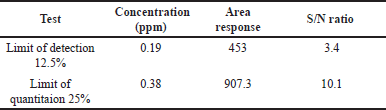 | Table 3. Detection limit and quantification limit results. [Click here to view] |
Linearity and range
In the process of method validation, we assess the method’s ability to generate accurate and proportionate analytical responses within a defined concentration range, thereby ensuring its suitability for quantitative analysis. We meticulously prepared standard solutions of methyl 2-chloro acetate at diverse concentrations spanning from the limit of detection to 120% (12.5%, 25%, 50%, 80%, 100%, and 120%) of the working level (1.5 ppm). Subsequently, we conducted chromatographic analyses on these solutions, as outlined in Table 4 and Figure 7, to ascertain the linearity and range of the method.
We calculated the correlation coefficient between the mean area response and the standard solution concentration. The correlation coefficient obtained from the linearity graph plotting the mean area response against concentration in ppm was 0.9996. The linearity extended from the LOQ (0.38 ppm) to the higher level (1.8 ppm) and the regression statistics for linearity were displayed in Table 5.
Accuracy (recovery)
We evaluate the method’s capability to accurately quantify and recover known analyte quantities, thereby demonstrating its precision and reliability in real-world sample analysis. For each level, we prepared three sample solutions by spiking a homogeneous chlordiazepoxide hydrochloride sample at concentrations of 25%, 50%, 100%, and 120%. We subsequently analyzed these solutions using the prescribed method and calculated the recovery rates to assess the procedure’s accuracy. The recovery rates ranged from 97.3% to 101.5% for solutions within 25% to 120% concentration range, confirming the method’s accuracy, as outlined in Table 6.
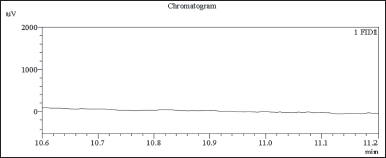 | Figure 4. GC-FID chromatogram diluent blank chromatogram, no interference observed around main peak retention time 10.8 minutes. [Click here to view] |
 | Figure 5. GC-FID chromatogram: MCA spiked sample (0.19 ppm LOD) [Click here to view] |
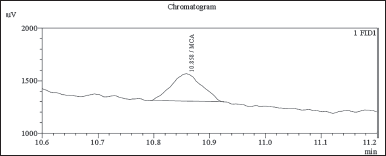 | Figure 6. GC-FID chromatogram: MCA spiked sample (0.38 ppm LOQ). [Click here to view] |
Finally, we conducted an indirect assessment of chloroacetyl chloride content by employing the developed and validated method on the chlordiazepoxide hydrochloride test samples. Chloroacetyl chloride was not detected in three batches of API samples as shown in Figures 10–12 and assay Table 13.
Robustness
We assessed the method’s robustness by evaluating its ability to consistently yield reliable results despite minor variations in experimental conditions, demonstrating its resilience and reliability in practical applications. Through a series of experiments, we demonstrated that this method maintains its robustness even when subjected to slight variations in gas chromatographic conditions. Throughout the experimental process, systematic alterations were implemented to optimize various instrumental parameters. Noteworthy adjustments included fluctuations in the carrier gas flow rate, ranging from 4.5 to 5.5 ml/min, with corresponding results meticulously documented in Table 7. In addition, subtle modifications were made to the detector temperature, demonstrating a range of variation from 220°C to 240°C, as systematically illustrated in Table 8. The injector temperature experienced adjustments within the interval of 135°C to 165°C, meticulously detailed in Table 9. Further refinements involved precise adjustments in the initial oven temperature, ranging from 36°C to 44°C, as systematically outlined in Table 10. Moreover, the temperature ramping rate underwent deliberate variation, oscillating between 8°C/min and 12°C/min, with methodical documentation provided in Table 11. These sequential and controlled changes were executed to assess their impact on the overall analytical performance and are fundamental to the refinement of the analytical methodology employed in this study. Under this diverse range of conditions, the tailing factor consistently stayed below 1.5, and the theoretical plate numbers consistently exceeded 100,000. In addition, we consistently observed peak area measurements with RSD below 5.0%.
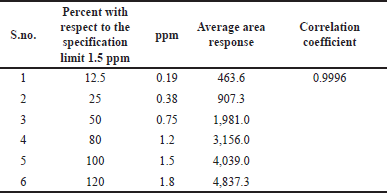 | Table 4. Linearity and the range of the method. [Click here to view] |
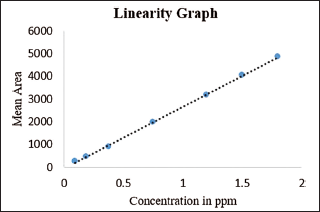 | Figure 7. Linearity graph mean area response versus concentration in ppm. [Click here to view] |
 | Table 5. Regression statistics for linearity. [Click here to view] |
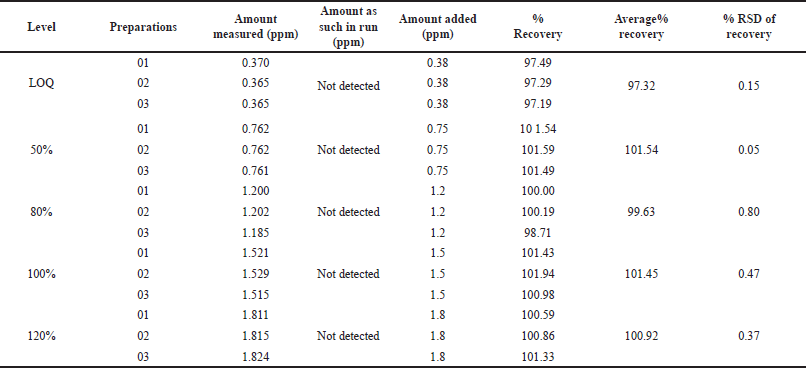 | Table 6. Accuracy of methyl-2- chloroacetate in chlordiazepoxide hydrochloride at different levels (n = 3). [Click here to view] |
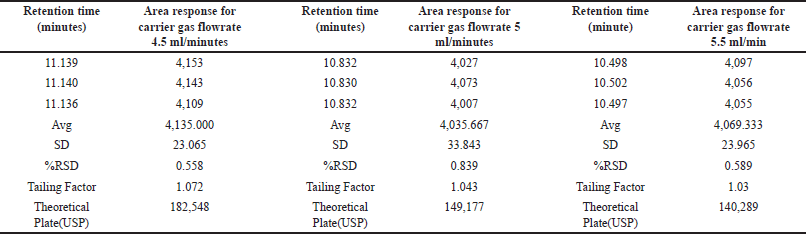 | Table 7. Robustness study: carrier gas flow rate variation (n = 3) versus area response of MCA, carrier gas flow rate (4.5 to 5.5 ml/minutes). [Click here to view] |
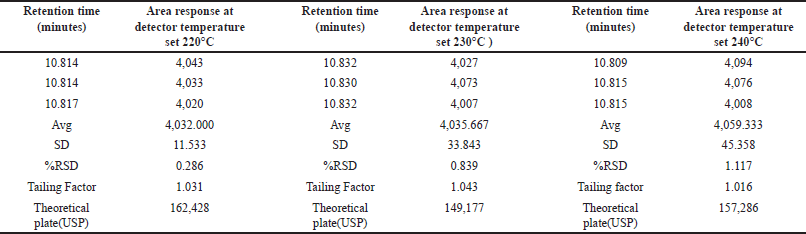 | Table 8. Robustness study: detector temperature variation (n = 3) versus area response of MCA (°C 220–240). [Click here to view] |
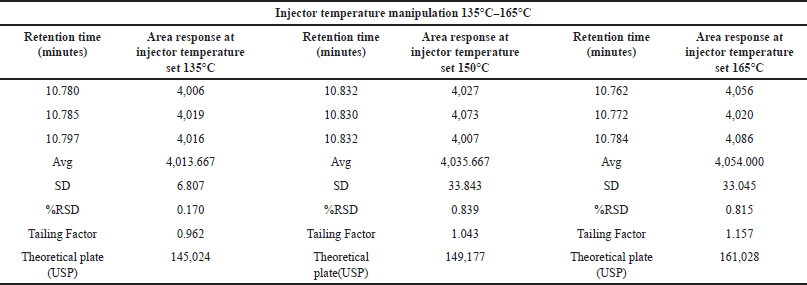 | Table 9. Robustness study: injector temperature variation (n = 3) versus area response of MCA (°C 135–165). [Click here to view] |
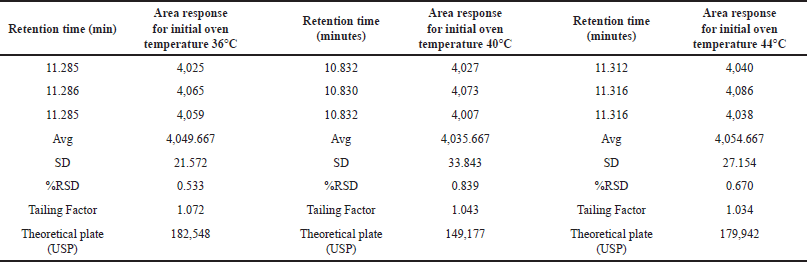 | Table 10. Robustness study: initial oven temperature variation (n = 3) versus area response of MCA (°C 36–44). [Click here to view] |
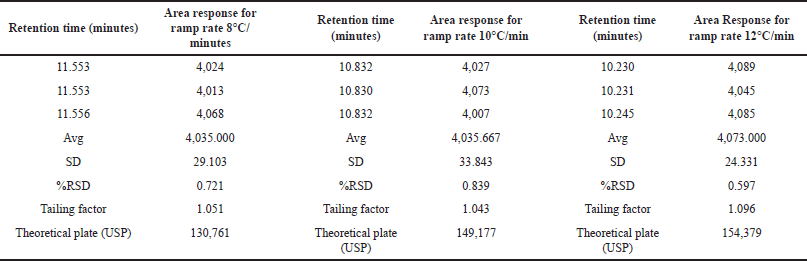 | Table 11. Robustness study: ramp rate (n = 3) versus area response of MCA (8°C/minutes to 12°C/minutes). [Click here to view] |
Solution stability
A solution maintains its chemical and physical properties, including component concentration and integrity, over a specified period under defined storage conditions, indicating its solution stability. To assess its stability over a 3-day period, we injected the standard solution of 1.5 ppm into the system, and the results in Table 12 demonstrate that the solution remained stable at room temperature (25°C) in clear glass throughout the entire duration.
 | Table 12. Solution stability results. [Click here to view] |
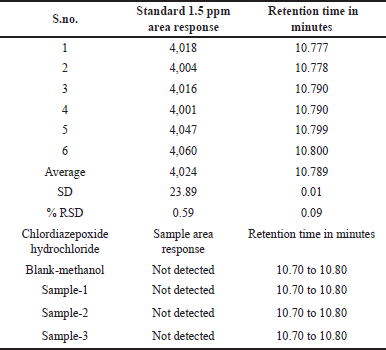 | Table 13. Assay of chloroacetyl chloride into chlordiazepoxide hydrochloride. [Click here to view] |
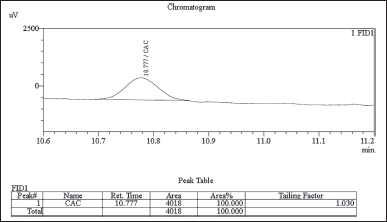 | Figure 8. GC-FID chromatogram: chloroacetyl chloroacetate standard 1.5 ppm spiked in chlordiazepoxide hydrochloride sample. [Click here to view] |
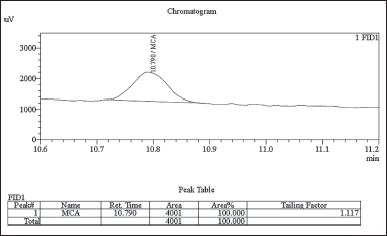 | Figure 9. GC-FID Chromatogram: MCA Standard 1.5 ppm spiked in chlordiazepoxide hydrochloride sample. [Click here to view] |
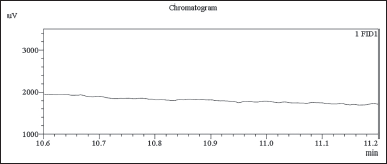 | Figure 10. GC-FID chromatogram: chlordiazepoxide hydrochloride API sample-01 in diluent blank. [Click here to view] |
 | Figure 11. GC-FID chromatogram: chlordiazepoxide hydrochloride API sample-02 in diluent blank. [Click here to view] |
 | Figure 12. GC-FID chromatogram: chlordiazepoxide hydrochloride API sample-03 in diluent blank. [Click here to view] |
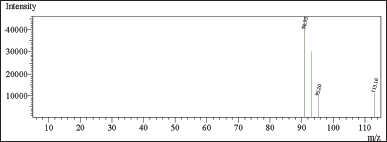 | Figure 13. Mass spectrum of CAC in dichloromethane [Click here to view] |
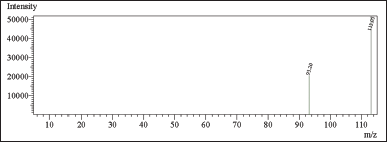 | Figure 14. Mass spectrum of CAC in ethylene dichloride. [Click here to view] |
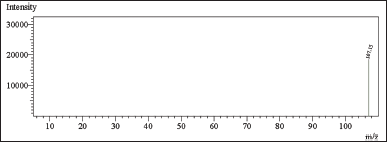 | Figure 15. Mass spectrum of CAC in methanol forming MCA at –ve polarity. [Click here to view] |
CONCLUSION
In summary, we have successfully developed a simple, expeditious, and secure (GC-FID) method for the quantitative determination of chloroacetyl chloride in chlordiazepoxide hydrochloride drug substance. This method has demonstrated exceptional precision and accuracy throughout its development and validation stages. In full compliance with the rigorous guidelines outlined by the International Conference on Harmonisation (ICH), our method has excelled in numerous critical analytical parameters, including specificity, system suitability, precision, detection and quantification limits, linearity, accuracy (recovery), robustness, and solution stability for three days.
The successful application of this method in the analysis of three distinct batches of chlordiazepoxide hydrochloride samples, all of which did not exhibit any trace of MCA, underscores the method’s reliability and suitability for use in the pharmaceutical industry. In upholding its commitment to ensuring patient safety, the pharmaceutical sector can confidently rely on this robust analytical approach to maintain the quality and integrity of the active drug substance.
LIST OF ABBREVIATIONS
CAC, Chloroacetyl chloride; GC-FID, Gas chromatography with flame ionization detector; ICH, The International Conference of Harmonization; MCA, Methyl 2-chloroacetate
ACKNOWLEDGMENTS
The authors wish to extend their sincere gratitude to REVA University for the invaluable support and resources provided during this research. Special recognition and appreciation are reserved for Professor Madhusudhana Reddy M.B., whose guidance and mentorship have been instrumental in the success of this endeavor.
Furthermore, the authors would like to express heartfelt thanks to Flow Chem Pharma Pvt. Ltd. and Trroy Life Sciences Pvt. Ltd., Bangalore, for their generous provision of laboratory facilities and essential support. These contributions played a significant role in advancing the research efforts and are greatly acknowledged.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Guo L, Zhang X, Zhou Z, Shi M, Jian X, Dong L. Case report: occupational poisoning incident from a leak of chloroacetyl chloride in Jinan, Shandong, China. Front Public Health. 2023;11:1215293.
2. Koenig G, Lohmar E, Rupprich N, Lison M, Gnass A. Chloroacetic acids. In Ullmann’s encyclopedia of industrial chemistry, 2012. CrossRef
3. Morris ED, Bost JC. Acetic acid, halogenated derivatives. In Kirk-Othmer encyclopedia of chemical technology. 2002. CrossRef
4. Steinnes O, Shen Q, Hagen K. Molecular structure and conformation of gaseous chloroacetyl chloride as determined by electron diffraction. J Mole Struct. 1980 May 1;64:217–28. CrossRef
5. Zheng X, Luo L, Zhou J, Ruan X, Liu W, Zheng F. Development and validation of a general derivatization HPLC method for the trace analysis of acyl chlorides in lipophilic drug substances. J Pharm Biomed Anal. 2017 Jun 5;140:327–33. CrossRef
6. Dolan DG, Naumann BD, Sargent EV, Maier A, Dourson M. Application of the threshold of toxicological concern concept to pharmaceutical manufacturing operations. Regul Toxicol Pharmacol. 2005 Oct 1;43(1):1–9. CrossRef
7. Delaney EJ. An impact analysis of the application of the threshold of toxicological concern concept to pharmaceuticals. Regul Toxicol Pharmacol. 2007 Nov 1;49(2):107–24. CrossRef
8. Dewhurst I, Renwick AG. Evaluation of the threshold of toxicological concern (TTC)–challenges and approaches. Regul Toxicol Pharmacol. 2013 Feb 1;65(1):168–77. CrossRef
9. Hennes EC. An overview of values for the threshold of toxicological concern. Toxicol Lett. 2012 Jun 20;211(3):296–303. CrossRef
10. Kroes R, Renwick AG, Cheeseman M, Kleiner J, Mangelsdorf I, Piersma A, et al. Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Chem Toxicol. 2004 Jan 1;42(1):65–83. CrossRef
11. Renwick AG. Structure-based thresholds of toxicological concern—guidance for application to substances present at low levels in the diet. Toxicol Appl Pharmacol. 2005 Sep 1;207(2):585–91. CrossRef
12. Snodin DJ. Genotoxic impurities: from structural alerts to qualification. Org Process Res Dev. 2010 Jul 16;14(4):960–76. CrossRef
13. Plošnik A, Vracko M, Sollner Dolenc M. Mutagenic and carcinogenic structural alerts and their mechanisms of action. Arhiv za higijenu rada i toksikologiju. 2016 Sep 22;67(3):169–82. CrossRef
14. Raillard SP, Bercu J, Baertschi SW, Riley CM. Prediction of drug degradation pathways leading to structural alerts for potential genotoxic impurities. Org Process Res Dev. 2010 Jul 16;14(4):1015–20. CrossRef
15. Sutter A, Amberg A, Boyer S, Brigo A, Contrera JF, Custer LL, et al. Use of in silico systems and expert knowledge for structure-based assessment of potentially mutagenic impurities. Regul Toxicol Pharm. 2013 Oct 1;67(1):39–52. CrossRef
16. Pradeep P, Judson R, DeMarini DM, Keshava N, Martin TM, Dean J, et al. An evaluation of existing QSAR models and structural alerts and development of new ensemble models for genotoxicity using a newly compiled experimental dataset. Comput Toxicol. 2021 May 1;18:100167. CrossRef
17. Benigni R, Bossa C. Structural alerts of mutagens and carcinogens. Current Comput-Aided Drug Design. 2006 Jun 1;2(2):169–76.
18. Dobo KL, Greene N, Cyr MO, Caron S, Ku WW. The application of structure-based assessment to support safety and chemistry diligence to manage genotoxic impurities in active pharmaceutical ingredients during drug development. Regul Toxicol Pharmacol. 2006 Apr 1;44(3):282–93. CrossRef
19. Enoch SJ, Cronin MT. Development of new structural alerts suitable for chemical category formation for assigning covalent and non-covalent mechanisms relevant to DNA binding. Mutat Res/Genet Toxicol Environ Mutagen. 2012 Mar 18;743(1–2):10–9. CrossRef
20. Munro IC, Renwick AG, Danielewska-Nikiel B. The threshold of toxicological concern (TTC) in risk assessment. Toxicol Lett. 2008 Aug 15;180(2):151–6. CrossRef
21. Müller L, Mauthe RJ, Riley CM, Andino MM, De Antonis D, Beels C, et al. A rationale for determining, testing, and controlling specific impurities in pharmaceuticals that possess potential for genotoxicity. Regul Toxicol Pharmacol. 2006 Apr 1;44(3):198–211. CrossRef
22. Ridings JE, Barratt MD, Cary R, Earnshaw CG, Eggington CE, Ellis MK, et al. Computer prediction of possible toxic action from chemical structure: an update on the DEREK system. Toxicology. 1996 Jan 8;106(1–3):267–79. CrossRef
23. Müller M, Birner G, Dekant W. Reactivity of haloketenes and halothioketenes with nucleobases: chemical characterization of reaction products. Chem Res Toxicol. 1998 May 18;11(5):454–63. CrossRef
24. ICH M7. Assessment And Control Of DNA Reactive (Mutagenic) Impurities In Pharmaceuticals To Limit Potential Carcinogenic Risk M7(R1) Current Step 4 Version Dated 31 March 2017.
25. Teasdale A. ICH M7: assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. In: Teasdale A, Elder D, Nims RW, editors. ICH quality guidelines: an implementation guide. Hoboken, NJ: Wiley; 2017 Sep 27. pp. 667–99.
26. Barber C, Amberg A, Custer L, Dobo KL, Glowienke S, Van Gompel J, et al. Establishing best practice in the application of expert review of mutagenicity under ICH M7. Regul Toxicol Pharmacol. 2015 Oct 1;73(1):367–77. CrossRef
27. Amberg A, Beilke L, Bercu J, Bower D, Brigo A, Cross KP, et al. Principles and procedures for implementation of ICH M7 recommended (Q) SAR analyses. Regul Toxicol Pharmacol. 2016 Jun 1;77:13–24. CrossRef
28. Khan M, Jayasree K, Reddy KK, Dubey PK. A validated CE method for determining dimethylsulfate a carcinogen and chloroacetyl chloride a potential genotoxin at trace levels in drug substances. J Pharm Biomed Analy. 2012;58:27–33. CrossRef
29. Morissette MF, Wigman L, Tso J. Trace level determination of chloroacetyl chloride and degradation products by derivatization gas chromatography. J Pharm Biomed Anal. 2018 Jan 30;148:93–9. CrossRef
30. Langvardt PW, Nestrick TJ, Hermann EA, Braun WH. Derivatization procedure for the determination of chloroacetyl chloride in air by electron capture gas chromatography. J Chromatogr A. 1978 Jun 11;153(2):443–50. CrossRef
31. Anerao A, Patil B, Solse V, Patil C. Quantification of genotoxic impurities in tadalafil by simple and sensitive gas chromatography technique. Anal Chem Lett. 2019 Jul 4;9(4):518–25. CrossRef
32. Kennedy D. Quantitative analysis of acid chlorides using automatic cold on-column injection. HRC and CC. J High Resol Chromatogr Chromatogr Commun. 1988;11(4):350–1. CrossRef
33. Klein AJ, Morrell SG, Hicks OH, Worley JW. Determination of chloroacetyl chloride in air by high-performance liquid chromatography. Anal Chem. 1986 Apr 1;58(4):753–5. CrossRef
34. McCullough PR, Worley JW. Sampling of chloroacetyl chloride in air on solid support and determination by ion chromatography. Anal Chem. 1979 Jul 1;51(8):1120–2. CrossRef
35. Zhou Y, Ni W, Liu Y, Huang P, Su M. Determination of potential genotoxic impurities chloroacetyl chloride and chloroacetic acid in azintamide raw material. J China Pharm Univ. 2022:300–5.
36. Langhorst ML. Monitoring airborne reactive chemicals by derivatization and high performance thin layer chromatography—anhydrides, acid halides, isocyanates. Am Indust Hygiene Assoc J. 1985 May 1;46(5):236–43. CrossRef
37. Liu DQ, Sun M, Kord AS. Recent advances in trace analysis of pharmaceutical genotoxic impurities. J Pharm Biomed Anal. 2010 Apr 6;51(5):999–1014. CrossRef
38. Robinson DI. Control of genotoxic impurities in active pharmaceutical ingredients: a review and perspective. Org Process ResDev. 2010 Jul 16;14(4):946–59. CrossRef
39. Jacobson-Kram D, McGovern T. Toxicological overview of impurities in pharmaceutical products. Adv Drug Deliv Rev. 2007 Jan 10;59(1):38–42. CrossRef
40. Raman NV, Prasad AV, Reddy KR. Strategies for the identification, control and determination of genotoxic impurities in drug substances: a pharmaceutical industry perspective. J Pharm Biomed Anal. 2011 Jun 25;55(4):662–7. CrossRef
41. Pierson DA, Olsen BA, Robbins DK, DeVries KM, Varie DL. Approaches to assessment, testing decisions, and analytical determination of genotoxic impurities in drug substances. Org Process Res Dev. 2009 Mar 20;13(2):285–91.
42. Marselos M, Vainio H. Carcinogenic properties of pharmaceutical agents evaluated in the IARC Monographs programme. Carcinogenesis. 1991 Oct 1;12(10):1751–66. CrossRef
43. Li Y, Yang H, Jin L, Wu WP, Yang D. Theoretical study on the mechanism of chloroacetyl chloride decomposition. Comput Theor Chem. 2016 Jun 15;1086:52–7. CrossRef
44. Bock H, Hirabayashi T, Mohmand S, Solouki B. Unstable intermediates in the gaseous phase: the thermal decomposition of acyl chlorides RCOCI. Angewandte Chemie Int Edit English. 1977 Feb;16(2):105–7.
45. ICH, Q2A (R1). Validation of analytical procedures: text and methodology. In: International Conference on Harmonization, Geneva, 2005. Vol. 1994, p. 17.