
INTRODUCTION
Drugs are substances used to treat, diagnose, cure, mitigate, treat, and prevent diseases, either as drugs or as constituents of drugs [1]. In the field of medicinal chemistry, natural compounds are extracted or new molecules are synthesized. The utilization of heterocyclic quinazolines is critical in the advancement of pharmaceuticals. Heterocyclic quinazolines are biologically active compounds with various properties such as anti-inflammatory, antibacterial, antifungal, anticonvulsant, anti-HIV, anticancer, analgesic, antidepressant, and antioxidant [2]. The exploration of quinazoline derivatives has yielded an extensive collection of compounds exhibiting a wide range of biological activities. The quinazoline nucleus has garnered considerable interest within the field of medicinal chemistry in recent times. One may reasonably hypothesize that it functioned as a pharmacophore element in the production of medically useful chemicals [2].
Quinazolines possess pharmacological effects including anti-inflammatory, anti-tuberculous, anticancer, and antiviral properties. A number of pharmaceutical compounds derived from quinazolines have received approval. Quinazolines and their derivatives exhibit anticancer properties by inhibiting DNA repair enzymes, EGFR, tubulins, apoptosis, and DNA replication and transcription. Notable examples include prazosin and doxazosin, which are approved for the treatment of benign prostatic hyperplasia and post-traumatic stress disorder, respectively; erlotinib and gefitinib, which are utilized in the treatment of pancreatic and lung cancer, respectively [3]. According to the World Health Organization, cancer is now the primary cause of death, exceeding both coronary heart disease and stroke. According to the International Agency for Cancer Research, it is projected that by 2040, the number of new cases and deaths caused by cancer will be twice as high as they are now [4]. Cancer cells, instead of being limited by typical limits on growth, acquire resistance and multiply as a result of their inherent drive. Moreover, they possess the capacity to spread beyond their original location by invading neighboring tissues and clustering in distinct areas of the body, which is an even more dangerous characteristic. Malignant tumors that are aggressive in nature have a prolonged course and may be lethal when they cause damage to vital organs and tissues in the whole body [5].
The role of growth factor-driven signaling in the development of human malignancy has been well established. The influence of growth factors and the corresponding transmembrane receptor tyrosine kinases on cell differentiation, survival, migration, and proliferation is substantial. A category of growth factors, comprising proteins resembling epidermal growth factor (EGF) and neuregulins, induces cellular division through the activation of EGF receptor (EGFR) family members, including HER2-4 and EGFR receptors [6,7]. This extensively conserved signaling module is vital to the morphogenesis of numerous organisms, from nematodes to humans, and has been associated with the growth and development of numerous types of human tumor cells. The EGFR family, which consists of over 30 ligands and four receptors, forms the foundation of an intricate, multilayered network that regulates human signaling [8,9].
The epidermal growth factor receptor (EGFR) is mutable, with the T790M mutation limiting the efficacy of anti-cancer agents owing to the emergence of drug resistance conferred by a second mutation [10]. Thr790Met, which is also known as T790M, is a mutation of the EGFR that may help us understand how cells go from being healthy to being cancerous. At position 790 of the exon, a threonine was changed to methionine, a crucial inhibitor specificity determinant in the ATP-binding pocket [11,12].To fathom drug action mechanisms, one must comprehend the binding forces between drugs and their receptors. More than 10 quinazoline derivatives with potent anti-carcinogenic properties have been discovered in the last decade. This article goes into great detail about different quinazoline chemical parts, how they work computationally, and how their structure affects their ability to bind to EGFR. To fathom drug action mechanisms, one must comprehend the binding forces between drugs and their receptors [13]. As far as we know, no such research based on interaction has been conducted. This article discusses the computational aspects of molecular interactions with EGFR and the ADMET (Absorption, distribution, metabolism, excretion, and toxicity) properties. The ADMET investigation was conducted using version 3.0 of the QikProp module of Schrodinger maestro [14].
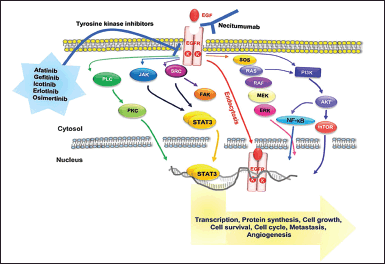 | Figure 1.Graphical representation of EGFR signal transduction and its targeted therapy. The anticancer drugs-containing medications confront the challenge of causing drug resistance [9]. [Click here to view] |
METHODOLOGY
Molecular docking and EGFR inhibitors
The utilization of molecular docking analysis has been employed to investigate the selected molecule based on an extensive review of the literature. The protein data bank (PDB) provided the X-ray crystal structures of EGFR (PDB ID: 4I23) and mutant EGFR (PDB ID: 6LUD), while the PubChem database (https://pubchem.ncbi.nlm.nih.gov) provided the chemical structure of quinazoline derivatives. Using prime, the protein preparation wizard in Epik version 3.4 processed the protein structure and prepared the crystal structure for protonation, general minimization, H-bond optimization, ionization, and filling in loop and side-chain gaps. The result of the docking analysis is depicted in Table 1.
RESULTS AND DISCUSSION
4-anilinoquinazoline derivatives
As highly selective and potent EGFR inhibitors, Weijie Hou et al. synthesized a series of 4-anilinoquinazoline derivatives with a benzamide moiety at the sixth position. According to the researchers [15], Compound 1 had the maximum affinity for EGFR. Docking of compound 1 (−7.951) with EGFR (PDB ID: 4I23) reveals a valid H-bond of Met793 and Ser720 with a nitrogen of the quinazoline nucleus and oxygen of the benzamide moiety, which fits the molecule into the EGFR binding socket; additionally, compound 1 reveals an H-bond with Lys728 and the oxygen of the morpholine substituent at the seventh position, as well as 2 halogen bonds of Thr790 and Leu788 with the -Cl, meta to the aniline at fourth position to the nucleus. This indicated the higher affinity of compound 1 for the EGFR protein.
During docking with the T790 mutant receptor (−7.528) (PDB ID: 6LUD), the compound displays the required H-bond of Met793 and three additional H-bonds. Met793 is linked to the nitrogen of the quinazoline nuclease, and the other H-bonds are located at Ser720 with the oxygen of the benzamide moiety, Asn842, and Asp855 with the nitrogen of the morpholine moiety. Additionally, the nitrogen of morpholine forms a salt bridge with Asp855 and a pi-cation binding with Phe723. The benzamide moiety exhibits pi-pi stacking with Phe723, which are the linkages that allow the molecule to adhere securely within the binding pockets.
AstraZeneca discovered 4-anilinoquinazoline analogue compound 2 Aurora Kinase Inhibitor with an IC50 of 0.1 M [16]. The docking investigation of compound 2 and EGFR reveals the significant H-bonding interaction with Met793 and nitrogen of the aniline group substituted at the fourth position of the quinazoline nuclease. In addition, the molecule fits into the binding sites created by the morpholine nitrogen salt bridges with Asp800 and Glu804. This compound 2 on docking with T790M mutated EGF receptor the necessary Met793 H-binding is showing with nitrogen of aniline group at the fourth position of quinazoline nuclease. Another H-bond of Lys716 with morpholine nitrogen is present.
4-arylamino-6-(furan-2-yl) derivative
Zhang et al. discovered structural and inhibitory similarities in a series of 4-arylamino-6-(furan-2-yl) quinazolines with the efficacious EGFR inhibitors lapatinib, gefitinib, and erlotinib. Compound 3 demonstrated significant anti-proliferation properties [17]. According to docking investigations, compound 3 forms a specific H-bond with Met793 and the nitrogen in the quinazoline’s nucleus. In addition, Thr854 and Asp855 form two H-bonds with the oxygen of the furan ring and the hydroxyl group of the methanolic side chain of the furan ring, as well as a halogen bond with Cys797 and -Cl, whereas compound 3 exhibits only hydrophobic interactions, indicates the enthalpy- entropy compensation at the docking site, upon docking with the T790M receptor.
Chalcone hybrids
As EGFR inhibitors, Hisham et al. created hybrids of quinazoline-4-one and chalcone. With an IC50 of 0.11 M, which is equal to erlotinib’s IC50 of 0.08 M, compound 6 from the synthesized analogs has extremely effective inhibitory activity [18]. According to docking research, a crucial interaction between a protein and its chemical is a hydrophobic H-bond formed by Met793 and the carbonyl oxygen atom at the p-position of the benzamide in the quinazoline nucleus’s second position. have an additional H-bond with Lys745 and the oxygen atom at the fourth position of quinazoline. While docking with the T790M receptor, the necessary linkage of H-bonding of Met793 with the carbonyl oxygen of chalcone moiety and an additional H-bond with Lys745 and oxygen of the methoxy group is present.
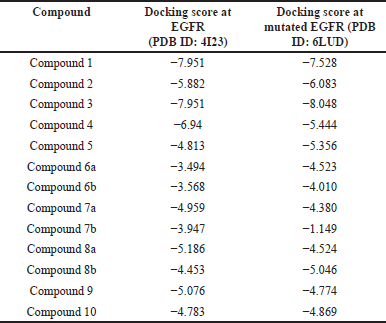 | Table 1. Docking score of chemical intermediates used in the analysis. [Click here to view] |
 | Figure 2. (a) Graphical representation of EGFR (b) Graphical representation of t790M mutated EGFR [10]. [Click here to view] |
6-bromo-2-(pyridine-3-yl)-4-substituted derivatives
Allam et al. created [19] EGFR inhibitors by synthesizing 6-bromo-2-(pyridine-3-yl)-4-substituted quinazoline analogs. Among these, unsubstituted benzothiazol-2-amine inhibits EGFR with an IC50 of 0.096 M [20]. A docking study of compound 5 with EGFR shows a relevant H-bond with Met793 and the nitrogen of the pyridine ring at the second position of quinazoline nuclease.
Thiourea containing derivatives
Sun and coworkers [19] created quinazoline compounds with thiourea that have VEGFR-2 and EGFR inhibitory activities. The thiourea containing quinazoline analogs compound 6a and 6b with the ether linkage at the 4-position, with an IC50 value of 0.02 M, and the thioether linked derivatives compound 7a and 7b, with an IC50 value of 0.01 M, are among those potent EGFR inhibitors [21]. Compound 6a shows the H-bonds with hydrophobic Met793 and nitrogen of quinazoline nuclease, which is the key interaction to show the EGFR inhibiting activity and two H-bonds of both nitrogen atoms in the thiourea moiety with Asn842 and Asp855. Also, compound 7b shows the necessary linkage of Met793 as dual with both the nitrogen atoms of thiourea moiety. Additionally, a halogen bond of bromine and Lys728 is present. During docking experiments, it was shown that these compounds interact with the T790M receptor of the EGFR by forming required hydrogen bonds with Met793. Additionally, additional hydrogen bonds are established between the thiourea group and the substituents on the phenyl ring.
Urea containing derivatives
Urea-based quinazoline derivatives were created by Abd El Hadi et al. [22] and are effective VEGFR-2 inhibitors. The amide derivative compound 8a had a methyl amide side chain at the second position of the quinazoline and a methoxy group at the meta position of the phenyl ring after the urea. It significantly suppressed VEGFR-2 with an IC50 value of 12.1 nM. Valid interactions between compound 8a and EGFR are revealed by a docking study. H-bonds exist with Met793 and the carbonyl oxygen of the methylamide side chain at the second position of quinazoline nuclease. Met793 and the -NH of aniline group of urea moiety at fourth position of quinazoline nuclease make the necessary H-bond in compound 8b. An H-bond of Lys728 and carbonyl oxygen of ester moiety at the second position of quinazoline nuclease is also made by the ester derivative compound 8b, which also has an IC50 of 14.8 Nm [22].
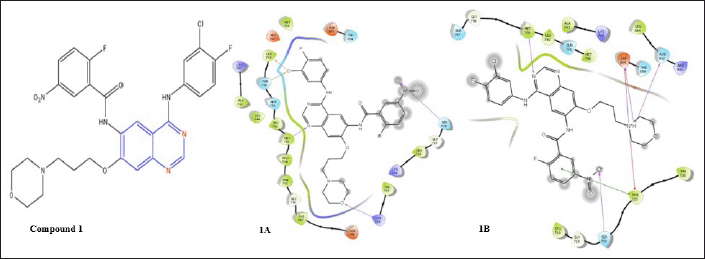 | Figure 3. Schematic of the molecular interactions of compound 1, N-(4-((3-chloro-4-fluorophenyl)amino)-7-(3-morpholinopropoxy)quinazolin-6-yl)-2-fluoro-5-nitrobenzamide on EGFR (1A) and on mutated EGFR (1B). [Click here to view] |
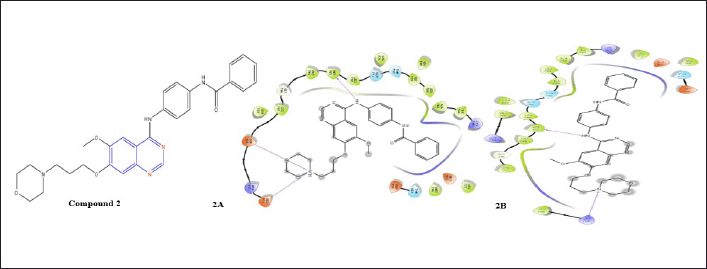 | Figure 4. Molecular interaction diagram of compound 2; N-(4-((6-methoxy-7-(3-morpholino propoxy)quinazolin-4-yl)amino)phenyl)benzamide on EGFR (2A) and on mutated EGFR (2B). [Click here to view] |
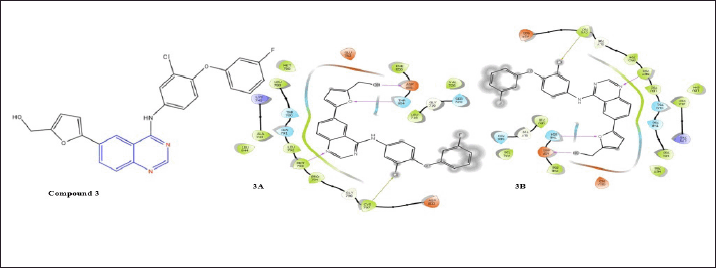 | Figure 5. Molecular interaction diagram of compound 3; (5-(4-((3-chloro-4-(3-fluorophenoxy) phenyl)amino)quinazolin-6-yl)furan-2-yl)methanol on EGFR (3A) and on mutated EGFR (3B). [Click here to view] |
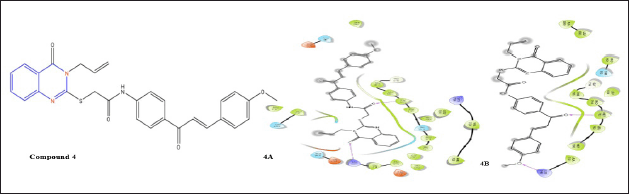 | Figure 6. Molecular interaction diagram of compound 4; (E)-2-((3-allyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(4-(4-(4-methoxyphenyl)buta-1,3-dien-2-yl)phenyl)acetamide on EGFR (4A) and on mutated EGFR (4B). [Click here to view] |
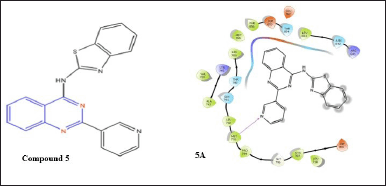 | Figure 7. Molecular interaction diagram of compound 5; N-(2-(pyridin-3-yl)quinazolin-4-yl)benzo[d]thiazol-2-amine on EGFR. [Click here to view] |
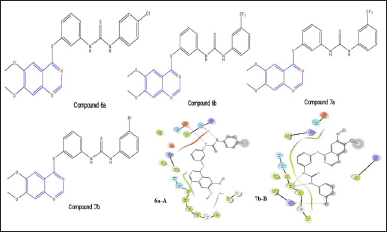 | Figure 8. Structure of compounds 6a, 6b, 7a, and 7b. 6a-A and 7b-B represents the molecular interaction diagram of compound 6a; 1-(4-chlorophenyl)-3-(3-((6,7-dimethoxyquinazolin-4-yl)oxy)phenyl)thiourea and compound 7b; 1-(3-bromophenyl)-3-(3-((6,7-dimethoxyquinazolin-4-yl)thio)phenyl)thiourea on EGFR. [Click here to view] |
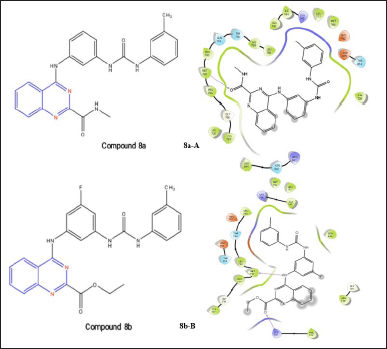 | Figure 9. Molecular interaction of compound 8a; N-methyl-4-((3-(3-(m-tolyl)ureido)phenyl)amino)quinazoline-2-carboxamide and compound 8b; 4-((3-fluoro-5-(3-(m-tolyl)ureido)phenyl)amino)-N-methylquinazoline-2-carboxamide (8a-A and 8b-B) on EGFR. [Click here to view] |
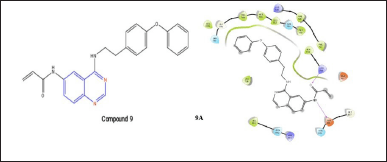 | Figure 10. Molecular interaction of compound 9; N-(4-((4-phenoxyphenethyl)amino)quinazolin-6-yl)acrylamide on mutated EGFR (9A). [Click here to view] |
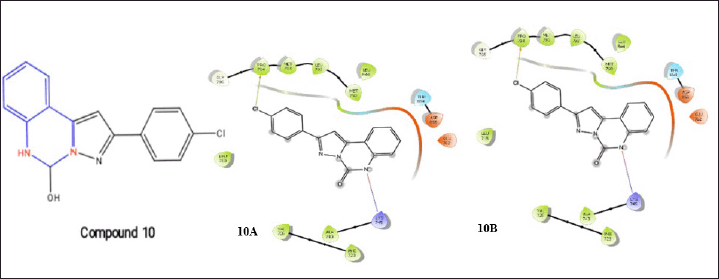 | Figure 11. Molecular interaction diagram of compound 10; 2-(4-chlorophenyl)-5,6-dihydropyrazolo[1,5-c]quinazolin-5-ol on EGFR (10A) and muted EGFR (10B). [Click here to view] |
Michael acceptor group derivatives
A quinazoline analog compound 9 with a Michael acceptor group at the 6-position was created by Shi et al. [6] and showed some CDK inhibition [16]. With the help of hydrophobic interactions, compound 9, and EGFR, this molecule fits into the binding socket during docking. Additionally, the oxygen of the benzamide group and Met793 create an H-bond, which is a crucial interaction for EGFR inhibitory activity. While docking with T790M the molecule fit into the binding pocket with two H-bonds of Lys745 and Asp855 and a side chain at the sixth position of quinazoline nuclease.
Dihydropyrazolo [1,5-C] derivative
Gagandeep Kaur et al. [23] developed dihydropyrazolo[1,5–c] quinazolines, which are catalytic inhibitors of human topoisomerase I. One of the derivatives, compound 10, demonstrated remarkable activity as evidenced by its IC50 value of 3.86 M [23]. Compound 10 forms a salt bridge with a nitrogen of quinazoline, indicating the stabilization of the complex, and Lys745 and one halogen bond with -Cl and Pro794, during its docking mechanisms with the EGFR and T790M receptors.
In cancer treatment, the EGFR plays a crucial function; it regulates the differentiation and proliferation of cancer cells within the body [24,25]. By selectively targeting the EGFR, it is possible to potentially cure the malignant condition. The primary challenge associated with anti-cancer medications is their susceptibility to drug resistance; thus, even the most potent EGFR inhibitors often experience a decline in efficacy [26]. The predominant mutation observed is T790M. This study assesses the EGFR-inhibiting activity of quinazoline scaffolds as well as their in-silico ADMET properties. In accordance with the EGFR inhibitory activity criterion, the ADMET properties of the molecule were selected. The compounds that have been presented exhibit the requisite molecular interactions with both the EGFR and T790M mutant receptors. These interactions have the potential to form the foundation for the development and production of innovative scaffolds that target cancer cell lines.
CONCLUSION
Computational modeling leverages the chemical molecular structure and its interaction with the EGFR to develop a therapeutic strategy and ultimately achieve cancer eradication. This review paves the way for a comprehensive understanding of clinical candidates, including their structural framework and interaction patterns, once more from the standpoint of computational learning. Cancer cells’ uncontrolled growth leads to invasiveness and lethal tumors. Growth factor-driven signaling influences malignancy, with neurotransmitters and proteins like EGF stimulating EGF receptor EGFR family members. The EGFR family, consisting of over 30 ligands and four receptors, forms a complex human signal transduction network. The T790M mutation in the EGFR kinase enhances treatment resistance by increasing the affinity for ATP. This study used a molecular design tool to evaluate quinazoline derivatives with the highest IC50 values against EGFR and analyze molecular interactions in the binding site to uncover novel compounds with promising potential. The knowledge acquired from this review could potentially inspire the development of novel frameworks or scaffolds that more efficiently target EGFR.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Silverman RB. The organic chemistry of drug design and drug action, power PDF. Amsterdam, The Netherlands: Elsevier; 2005 Feb 4.
2. Gomaa HA. A comprehensive review of recent advances in the biological activities of quinazolines. Chem Biol Drug Design. 2022 Nov; 100(5):639–55.
3. Mishra S. Quinazolinone and quinazoline derivatives: synthesis and biological application. Quinazolinone and quinazoline derivatives. IntechOpen. 2020 May 6;10:75–90.
4. Mattiuzzi C, Lippi G. Cancer statistics: a comparison between world health organization (WHO) and global burden of disease (GBD). Eur J Public Health. 2020 Oct; 30(5):1026–7.
5. Patergnani S, Danese A, Bouhamida E, Aguiari G, Previati M, Pinton P, et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int J Mol Sci. 2020 Nov 6;21(21):8323.
6. Shin I. HER2 Signaling in breast cancer. In translational research in breast cancer. Singapore: Springer Singapore; 2021. pp 53–79.
7. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004 Jun 1;59(2):S21–6.
8. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006 Jan 17;366(1):2–16.
9. Kim M, Baek M, Kim DJ. Protein tyrosine signaling and its potential therapeutic implications in carcinogenesis. Curr Pharm Design. 2017 Aug 1;23(29):4226–46.
10. Gao X, Le X, Costa DB. The safety and efficacy of osimertinib for the treatment of EGFR T790M mutation positive non-small-cell lung cancer. Expert Rev Anticancer Ther. 2016 Apr 2;16(4):383–90.
11. Ko B, Paucar D, Halmos B. EGFR T790M: revealing the secrets of a gatekeeper. Lung Cancer: Targets Ther. 2017 Oct 9:147–59.
12. Khrustalev VV, Khrustaleva TA, Poboinev VV, Yurchenko KV. Mutational pressure and natural selection in epidermal growth factor receptor gene during germline and somatic mutagenesis in cancer cells. Mutat Res. 2019 May 1;815:1–9.
13. Kang JJ, Ko A, Kil SH, Clair JM, Shin DS, Wang MB, et al. EGFR pathway targeting drugs in head and neck cancer in the era of immunotherapy. Biochim Biophys Acta Rev Cancer. 2023 Jan 1;1878(1):188827.
14. Thangaraj K, Arumugasamy K, Natesan K, Ramasamy S, Cyril R, Singh SK, et al. In Silico molecular docking analysis of Orientin, a potent glycoside of luteolin against BCL-2 family proteins. J. Chem. Pharm. Res. 2017;9:65–72.
15. Mohammed AH, Gomaa RM, El-Sayed MA, Selim KB. Synthesis, antitumor activities, and molecular modeling of 4-anilinoquinazoline derivatives as EGFR-TK inhibitors. Pharm Chem J. 2024;6:1–5.
16. Zayed MF. Medicinal chemistry of quinazolines as anticancer agents targeting tyrosine kinases. Sci Pharm. 2023 Mar 28;91(2):18.
17. Zhang Y, Zhang Y, Liu J, Chen L, Zhao L, Li B, et al. Synthesis and in vitro biological evaluation of novel quinazoline derivatives. Bioorg Med Chem Lett. 2017 Apr 1;27(7):1584–7.
18. Hisham M, Hassan HA, Gomaa HA, Youssif BG, Hayallah AM, Abdel-Aziz M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J Mol Struct. 2022 Apr 15;1254:132422.
19. Allam HA, Ibrahim BT, El-Dydamony NM, Fouad MA, Mohammed ER. Exploring new quinazolin-4 (3H)-one derivatives as CDK2 inhibitors: design, synthesis, and anticancer evaluation. Drug Dev Res. 2024 Apr;85(2):e22163.
20. Kaur J, Kaur S, Anand A, Islam N, Singh S, Singh A. An updated overview on the synthesis and anticancer evaluation of quinazoline derivatives. ChemistrySelect. 2023 Aug 11;8(30):e202302778.
21. Bansal R, Malhotra A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur J Med Chem. 2021 Feb 5;211:113016.
22. Abd El Hadi SR, Lasheen DS, Soliman DH, Elrazaz EZ, Abouzid KA. Scaffold hopping and redesign approaches for quinazoline based urea derivatives as potent VEGFR-2 inhibitors. Bioorg Chem. 2020 Aug 1;101:103961.
23. Kaur G, Cholia RP, Joshi G, Amrutkar SM, Kalra S, Mantha AK, et al. Anticancer activity of dihydropyrazolo [1, 5-c] quinazolines against rat C6 glioma cells via inhibition of topoisomerase II. Archiv der Pharmazie. 2018 Jun; 351(6):1800023.
24. Uribe ML, Marrocco I, Yarden Y. EGFR in cancer: signaling mechanisms, drugs, and acquired resistance. Cancers. 2021 Jun 1;13(11):2748.
25. Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nature Rev Cancer. 2021 Mar; 21(3):181–97.
26. Singh D, Attri BK, Gill RK, Bariwal J. Review on EGFR inhibitors: critical updates. Mini Rev Med Chem. 2016 Sep 1;16(14):1134–66.