
INTRODUCTION
The COVID-19 epidemic has emerged as a global extreme and has affected the global population. The global epidemic has broken out with 770,437,327 confirmed cases including 6,956,900 deaths as of September 6, 2023 [1]. The structure, mode of infection, and propagation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, have been well described [2]. The first step of infection is the attachment of the receptor binding domain (RBD) in the spike protein (SP) to the host cell receptor angiotensin converting enzyme-2 (ACE2). SP activation is mediated by transmembrane protease serine-2, a type 2 transmembrane-bound serine protease, and virus entry triggers RNA release followed by polyprotein translation [3]. Two important proteases, main protease (Mpro) and papain-like protease (PLpro) mediate the cleavage of polyproteins into functional nonstructural proteins (NSPs) [4]. The enzyme RNA-dependent RNA polymerase (RdRp) has a key role in viral replication and recapitulation [5]. It may be an attractive target for drug discovery and vaccine development against COVID-19. SARS-CoV-2 is a rapidly mutating virus that has given rise to several new strains of the virus, and therefore, the effectiveness of vaccination has become debatable. Since ancient times, herbal medicines have been used to treat viral diseases and the potential of some such medicines has been experimentally demonstrated [6]. Plant-derived molecules have evolved through repeated testing and reconditioning within a living system, and such molecules induce less toxicity, also several such molecules have shown diverse therapeutic activity due to their simultaneous effects on multiple targets. The importance of the multitarget activity of chemical molecules of plant origin has been well evaluated [7,8]. Recently, the common practice of “one disease–one target–one drug” has been transformed into a polypharmacological or multitarget approach to drug discovery as a potential solution to diseases of complex etiology and drug resistance problems [9].
Garcinia gummi-gatta (L.) Robs., also known as Malabar tamarind, is a tropical species under the genus Garcinia native to Southeast Asia and used against viral diseases in Indian traditional drug systems. In Indian Ayurvedic medicine, it is used to treat diarrhea, dysentery, and cancer. In addition, its anti-inflammatory, antimicrobial, anticancer, antihistaminic, antiobesity, antiadipogenic, hypolipidemic, vasodilating, and gastroprotective effects have been demonstrated [10]. Docking followed by lead optimization through analysis of physicochemical properties, drug-likeness, and absorption distribution metabolism and excretion (ADMET) properties using computational tools is the best way to rapidly primary confirmation of the medicinal properties of phytochemicals without large economic inputs. In light of the above reasons, phytochemicals from G. gummi-gatta were docked with selected SARS-CoV-2 and human targets, and lead molecules were identified using in silico methods.
MATERIALS AND METHODS
Selection and preparation of drug targets
The first selected target protein was the spike RBD of SARS-CoV-2 protein data bank (PDB ID 6M0J; chain B) with a resolution of 2.4 Å. The second is the ACE2 receptor, angiotensin-converting enzyme-related carboxypeptidase (PDB ID 1R42; chain A) at 2.20 Å resolution and the third is the Mpro (PDB ID 7BUY; chain A) at 1.60 Å resolution. Followed by SARS-CoV-2 RdRp (PDB ID 7BV2; strand A) at 2.5 Å resolution and the fifth target human nuclear factor-κB (NF-κB) (PDB ID 1IKN; strand A) at 2.30 Å. Active site residues were predicted using PDBSum. Crystal structures of the target proteins were obtained from the PDB database and initial processing was performed using AutoDock Tools. Before docking water molecules and heteroatoms were removed from all the targets and polar hydrogen and Gastieger charges were added to the aforementioned macromolecules [11].
Selection and preparation of ligands
Ninety-seven compounds from G. gummi-gutta were screened against five selected targets to identify the lead molecule. The 3-D structures of the phytochemicals and the control drug were downloaded from the PubChem database in .sdf format. Energy minimization was performed using Universal Force Field (uff) with conjugate gradient as the optimization algorithm. Ligands were converted to pdbqt file format using Open Babel within PyRx 0.8 [12].
Molecular docking
Docking of the selected targets and phytochemicals was performed using the AutoDock Vina tool, which follows the Lamarckian Genetic Algorithm [13], and it was accessed via the PyRx virtual screening software package. During molecular docking, the target proteins were considered rigid, and the phytochemicals (ligands) as flexible molecules. The grid boxes were laid out in such a way as to include the active site residues of each target in the grid box. For SP and ACE2, the X, Y, and Z coordinates were 43.2751, 55.1173, 30.4412; 25,000, 25,000, and 25,0000. Similarly, for Mpro, RdRp, and NF-κB, the X, Y, and Z coordinates were 25.2024, 40.5658, 40.0652; 25.0000, 25.0000, 30.3256; 85.2286, 65.2258, and 85.7493, respectively, with an exhaustiveness parameter of 8.
Postdocking analysis
Protein-ligand interaction was analyzed and visualized using PyMol 4.6.0 [14] and Discovery Studio Visualizer [15].
Physio-chemical and pharmacokinetic analysis
Pharmacokinetic properties such as absorption, distribution, metabolism, and elimination analysis of the top five results from each target were performed using the SwissADME online program [16] where molecular descriptors were calculated such as molecular weight, hydrogen bond donor, hydrogen bond acceptor, logP, and violation of Lipinski’s rule of five, further toxic properties were analyzed using pkCSM [17].
Molecular dynamic (MD) simulation
MD simulation is one of the powerful computer simulation techniques used to study the stability and dynamics of protein-ligand complex interactions. In the present study, we have performed MD simulation of the Mpro-lead complex and Mpro complexed with native inhibitor using Desmond for a time interval of 100 ns with the optimized potentials for liquid simulations all-atom force field. The docked complex was centered in the octahedron box and the system was solvated by a simple H2O point charge model with buffers at a distance of 10 Å around the protein. Overlapping water molecules were also removed and Na+ counter ions were added to neutralize the system. The number of atoms, pressure and temperature held constant system was achieved using a Nose-Hoover thermostat and a Martyne-Tobias-Klein barostat.
RESULTS AND DISCUSSION
Selection of the target proteins
The RBD in SP of SARS-CoV-2 forms the interface between SP and ACE2. The RBD binds to ACE2 with affinity in the low nanomolar range, suggesting its key functional role in binding with ACE2, and thus these two proteins are considered the most promising targets against viral entry [18]. The crystal structure of RBD is a 97.14 kDa glycoprotein with 832 amino acids and a five-stranded antiparallel β sheet (β1- β3- β5- β4- β2) core and β4- β5 sheets together form a receptor binding site known as the receptor binding motif, which directly interacts with ACE2. The crystal structure of ACE2 is a 76.98 kDa carboxypeptidase consisting of 673 amino acids [19]. Mpro or 3C-like protease has a key role in viral replication; cleaves polyproteins pp1a and pp1ab into 12 functional proteins. It is a 34.36 kDa homodimer with three domains each (domains I, II, and III). The substrate binding site is located in the cleft between domains I and II and contains catalytic dyads His41 and Cys145 that aid in substrate recognition [20]. Structural and functional annotation of SARS-CoV-2 Mpro and its role as a therapeutic target are summarized [21]. RdRp is an enzyme that inhibits viral RNA synthesis, and its structural and functional annotations have been reported [22]. It is a 159.19 kDa multisubunit replicase complex with Nsp12-Nsp7-Nsp8 subunits that has a pivotal role in viral replication. The increase in pro-inflammatory cytokine production and subsequent high death rate in patients with COVID-19 is modulated by the NF-κB signaling pathway [23]. The crystal structure of NF-kappa B is a 72.77 kDa transcription factor with 612 amino acid residues. NF-kappa B is a potential drug target for severe cases of COVID-19 [24].
The active site residues of selected target proteins determined using PDBSum were as follows: SP–Lys417, Gly446, Tyr449, Asn487, Tyr489, Gln493, Thr500, Asn501, and Tyr505; ACE2–Arg273, His345, Thr371, Glu375, His378, Glu402, Phe504, His505, Tyr515, Tyr510, and Arg514; and Mpro–Thr26, His41, Met49, Gly143, Ser144, Cys145, His164, Met165, Asp187, RdRp-Arg555, Asp623, Ser682, Thr687, and Asp760.
Selection of the ligands
Compounds of plant origin have shown multitarget inhibitory activity and such molecules induce fewer side effects compared to synthetic drugs [25]. For example, Keerthi et al. [26] screened 249 phytochemicals from Syzygium aromaticum (clove) against four targets of SARS-CoV-2, namely SP, ACE2, major protease (Mpro), and RdRp by an in silico method and found that 46 phytochemicals have an inhibitory effect on all the above-mentioned targets. Similarly, the plant-derived compounds β-sitosterol and campesterol have inhibitory activity on nine Russell viper venom proteins [27]. In addition, several plant-derived products used in modern medicine have multipurpose activity and are prescribed as drugs against many diseases [28]. From this perspective, a total of 97 phytochemicals from G. gummi-gutta were prepared for docking.
Molecular docking
The selected 97 phytochemicals from G. gummi-gutta were docked to each of the five selected targets, namely SP, ACE2, Mpro, RdRp, and NF-κB. Docked molecules with minimum binding energy ≤−6 kcal/mol were considered as active/hit molecules as reported earlier [29]. Of the 97 tested phytomolecules, 18 of them have significant activity on all the above-mentioned targets (Table 1). Out of 18 common hits, 8 of them, namely 1,4-dicaffeoylquinic acid, amentoflavone, cambogic acid, dihydromorelloflavone, luteolin-7-glucuronide, morelloflavone, oxyguttiferon K, and rheediaxanthon A have binding free energies/mol of −7 kcal/mol against all five targets (Supplementary Tables 1–4). Against the SP, only the compound amentoflavone showed H-bond interaction with the active site residues, GLN493 and ASN501, while 1,4-dicaffeoylquinic acid, dihydromorelloflavone, luteolin-7-glucuronide, camboic acid, rheediaxanthone A, and morelloflavone showed H-bond interaction with SP without the involvement of active site residues. Against ACE2, the compounds amentoflavone, cambogic acid, and luteolin-7-glucuronide formed H-bond interactions with the active residue ARG514, while oxyguttiferon K has H-bonds with the active residues ARG273 and HIS345. Similarly, dihydromorelloflavone has three H-bonds and out of eight hydrophobic interactions, two are with the active residues ARG273 and HIS345. The compound morelloflavone showed no H-bond with ACE2 and showed strong hydrophobic interactions, but none with the active site residue.
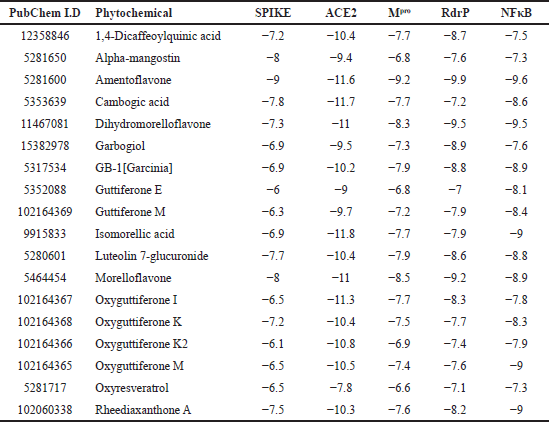 | Table 1. The docked results showing phytochemicals from G. gummi-gutta having free energy of binding ≤−6 kcal/mol against all the selected targets. [Click here to view] |
Docking interaction between Mpro with selected hits revealed that they all have H-bonding and hydrophobic interactions. The amentoflavone compound has the lowest binding energy and formed H-bonds with active residues HIS41, SER144, and CYS145. The second lowest binding energy with Mpro was recorded for morelloflavone, which has three H-bonds with active residues GLY143 and SER144. The third smallest binding energy was shown by dihydromorelloflavone, which showed four H-bonds and three hydrophobic interactions, but these interactions were not with any of the active residues. All five other compounds showed binding energy at the same level as the others and formed H-bonds with active site residues (Supplementary Table 1). The target RdRp showed both H-bonding and hydrophobic interactions with the selected eight hits, but only cambogic acid and luteolin-7-glucuronide showed H-bonding interaction with active residues. The former showed H-bonding with THR687 and the latter with ASP760. The NFkB target showed both H-bonding and hydrophobic interactions, but none of the selected hits interacted with its active residues.
The amentoflavone compound showed the lowest docking score against all targets (−9, −9.2, −9.9, and −9.6 kcal/mol against SP, Mpro, RdRp, and NF-κB) and formed H-bonds and hydrophobic interactions with all targets and interactions with active residues of three targets. The compound isomorelic acid with ACE2 showed the highest docking score (−11.8 kcal/mol). However, the docking scores of cambogic acid (−11.7 kcal/mol), amentoflavone (−11.6 kcal/mol), oxyguttiferon I (−11.3 kcal/mol), dihydromorelloflavone (−11 kcal/mol), and morelloflavone (−11 kcal/mol) were at the same level. The compound amentoflavone showed the lowest score for four of the five target drugs (Table 1). Therefore, it was chosen as the lead molecule. The docking interaction of amentoflavone with each of the selected targets is shown in Figure 1.
Physio-chemical and pharmacokinetic analysis
Physicochemical properties showed that except for rheediaxanthone A, all others violated Lipinski’s rule of five. The compounds 1,4-dicaffeoylquinic acid, dihydromorelloflavone, and morelloflavone showed three violations. However, with the exception of rheediaxanthone A, all others showed favorable drug similarity scores. Dihydromorelloflavone showed the highest drug-likeness score followed by luteolin 7-glucuronide and oxyguttiferon K, cambogic acid, 1,4-dicaffeoylquinic acid, morelloflavone, and amentoflavone.
ADMET analysis
The analysis of absorption parameters showed that all selected active compounds have water solubility in the range of −4.5 to −2.89 log mol/l. The compounds 1,4-dicaffeoylquinic acid, alpha-mangostin, dihydromorelloflavone, and luteolin-7-glucuronide showed lower Caco2 permeability values, corresponding to lower intestinal absorption. Among the hits, only 1,4-dicaffeoylquinic acid and luteolin-7-glucuronide showed intestinal absorption >30%, indicating a poor rate of absorption, while all others showed 70%–100%, indicating a high rate of intestinal absorption. P-glycoprotein, an ATP-binding cassette transporter, has an important function in drug availability, determining the influx and efflux of drugs from the system. All hits are P-glycoprotein substrates; four compounds, namely 1,4-dicaffeoylquinic acid, garbogiol, luteolin-7-glucuronide, and oxyresveratrol, do not act as inhibitors of P-glycoprotein I and II, while cambogic acid and isomorelic acid can only inhibit P-glycoprotein I. All compounds showed logKp >−2.5, which indicates good skin permeability. The distribution parameters showed that except for the compounds 1,4-dicaffeoylquinic acid and luteolin-7-glucuronide, all others showed a poor volume of distribution (VDss) as the VDss values were below 0.71 l/kg. Blood-brain barriers (BBBs) block the passage of drugs into the brain. If the logBB value of a compound is >0.3, it will easily cross the BBB, while if the logBB value is <−1, it will be poorly distributed to the brain. Only two compounds, oxyguttiferon I and oxyguttiferon M, have a logBB greater than 0.3, while cambogic acid, garbogiol, isomorelic acid, oxyguttiferon K2, oxyresveratrol, and rheediaxanthon A have a logBB less than −1. Compounds with logPS > −2 can enter the central nervous system (CNS), while compounds with logPS < −3 cannot enter the CNS. Among the hits, the compounds 1,4-dicaffeoylquinic acid, amentoflavone, dihydromorelloflavone, GB-1[garcinia], luteolin 7-glucuronide, and morelloflavone have logPS <−3 and the compounds alpha-mangostin, guttiferon F, oxyguttiferon I, oxyguttiferon K2, oxyguttiferon M, and rheediaxanthon A have logPS >−2; therefore, these molecules can penetrate the CNS. The remaining five compounds may have moderate CNC penetration. Cytochrome P450 (CYP) and hemoproteins play an important role in drug metabolism and blocking its activity has led to drug interactions when drugs are co-administered by increasing their plasma concentration and toxicity, resulting in adverse side effects and treatment failure. None of the compounds act as CYP2D6 substrates, while except for two compounds, garbodiol, and luteolin-7-glucuronide, all others are CYP3A4 substrates and except for guttiferon F, guttiferon M, oxyguttiferon I, oxyguttiferon K2, oxyguttiferon M, and rheediaxanthone A, all others are not CYP3A4 inhibitors. In the elimination profile, total clearance indicates the ability of the body to eliminate the drug from the system and is related to the bioavailability of drugs. The compounds alpha-mangostin, amentoflavone, garbogiol, guttiferon F, guttiferon M, luteolin 7-glucuronide, and oxyresveratrol showed a high overall clearance rate compared to all other hit molecules. Furthermore, none of the compounds act as renal Oct2 substrates. In the toxicity profile, Ames toxicity was noted for alpha-mangostin and garbodiol, hepatotoxicity was noted for oxyguttiferon I, oxyguttiferon K2, oxyguttiferon M, and rheediaxanthone A, and none of the compounds showed skin sensitization, Tetrahymena pyriformis and minnow toxicity.
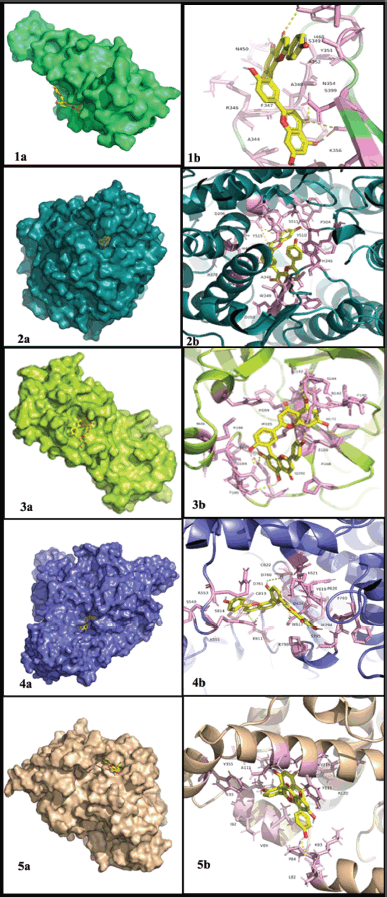 | Figure 1. Docking between the selected target proteins and the best lead amentoflavone (a) 3-D view and (b) 2-D view. 1) SP and amentoflavone, 2) ACE2 and amentoflavone, 3) Mpro and amentoflavone, 4) RdRp and amentoflavone, and 5) NF-κB and amentoflavone. [Click here to view] |
Because the compound amentoflavone has the lowest binding score against four out of five target proteins with good interactions involving active site residues and also showed favorable drug similarity and ADMET scores. Therefore, it is selected for further study.
MD simulation
MD simulation is an attractive computational application for analyzing the time-dependent stability of protein-ligand complexes through various parameters [30]. Therefore, an all-atom MD simulation was performed for two complexes, i.e., Mpro-lead complex and Mpro-inhibitor complex. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) parameters were used to analyze the stability of the protein-ligand complex during 100 ns of simulations. For the ligand in the Mpro-lead complex, the RMSD value was noted to be 1.2–2.7 Å during the simulation whereas the RMSD value was 0.4–2.8 Å in the Mpro-inhibitor complex. Overall, the RMSD plot shows very slight structural variations, indicating better stability of the protein-lead complex compared to the protein-inhibitor complex. However, there is no specific rule for the RMSD value to evaluate the consistency of a protein-ligand complex, as the RMSD value will depend on the size of the interacting molecule. RMSD is classified into three categories (a) good solutions when RMSD ≤2.0 Å, (b) acceptable solutions when RMSD is between 2.0 and 3.0 Å, and (c) poor solutions when RMSD ≥3.0 Å. When a protein-ligand pose is classified as good, it means that the scoring function reproduced the crystallographic binding orientation [31]. The average RMSD values of the protein backbone and the ligand in the Mpro-lead complex were 2.03 and 1.19 Å, thus indicating good structural reconstruction duration in the simulation of the protein-ligand complex. The structural integrity and controllability of each residue in the protein-ligand complex were analyzed using RMSF. The RMSF for the protein-main chain and side chain were 0.99 and 0.89 Å in the given protein-lead complex. For the Mpro-lead complex, fluctuations were observed at regions from residue indexes of 30–70, 140–200, 230–240, and 270–280. For inhibitor-bound Mpro complex, fluctuations were observed around regions 15–20, 30–40, 0–60, 145–160, and 180–190. Notably, most of the active site residues fall within these windows, favoring ligand interaction. Thus, low values of RMSD and RMSF indicate minor structural modifications and conformational changes. The simulation results are depicted in Figure 2a–d.
The protein–ligand interactions observed during a 100 ns simulation period are shown in Figure 3. Protein-ligand interactions can be divided into four types, i.e., hydrogen bonds, hydrophobic interactions, and ionic and water bridges. Stacked bar graphs are analyzed over the course of the trajectory, i.e., a value of 0.8 on the y-axis indicates that the interaction is maintained for 80% of the simulation time. In addition, a value above 0.1 indicates multiple types of interaction with the same residue. Here, Glu166 of the Mpro shows interaction with lead for 100% of the simulation time through water-bridged hydrogen bonds, and Gln189 interacts with lead for about 80% of the simulation time. Interaction analysis between Mpro and lead during 100 ns simulation shows that amino acids Ser46, Leu141, Gly143, Ser144, Cys145, Glu166, Asp187, Gln189, and Thr190 formed hydrogen bond interactions. Similarly, Met49 and Met165 form hydrophobic interactions. Notably, all active site residues show good interaction with the lead molecule, of which His41, Met49, Gly143, Cys145, His164, and Met165 interact through hydrogen bonds. The H-bond interaction plays a key role in protein-ligand interaction due to its strong influence on drug adsorption, specificity, distribution, and metabolism.
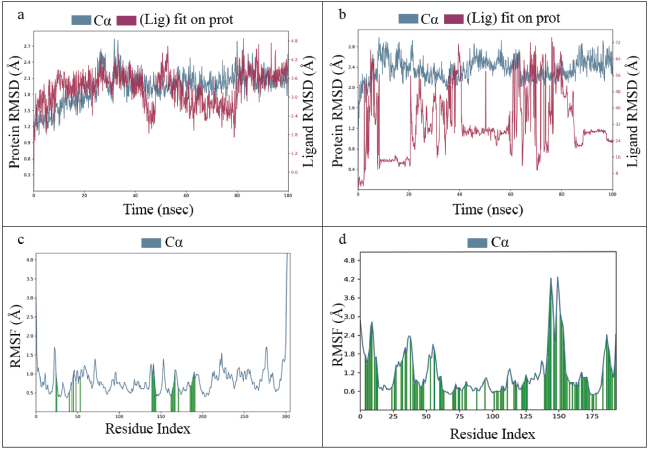 | Figure 2. a) RMSD of Mpro-lead complex, b) RMSD of Mpro-inhibitor complex, c) RMSF of Mpro-lead complex, and d) RMSF of Mpro-inhibitor complex. [Click here to view] |
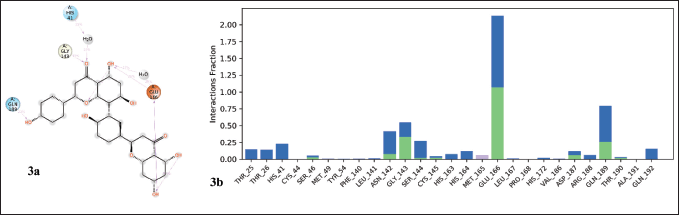 | Figure 3. Properties of ligand interaction with the protein. a) 2-dimensional interaction of lead molecule with protein and b) interaction of protein residues with the lead molecule. [Click here to view] |
The interaction between the spike glycoprotein of SARS-CoV-2 and human ACE2 is the primary step for the entry of the virus into the human body and its mechanism is described [32]. Inhibitors of these two targets can prevent the infection. Once the viral RNA enters the cell, it encodes two overlapping open reading frames (ORF1a and ORF1ab), which are translated into two large polyproteins (pp1a and pp1ab). These polyproteins are further processed by Mpro and PLpro to produce 16 NSPs. The NSP4-NSP16, released by Mpro cleavage, is responsible for viral genome replication and transcription. Blocking the activity of Mpro leads to the termination of viral RNA replication and transcription [33]. Similarly, the enzyme RdRp is inevitable for RNA replication, and its inhibitors can prevent viral replication [5]. The SARS-CoV-2 infection may induce an uncontrolled inflammatory response due to the abnormal functioning of NF-kB that leads to disease progression and subsequent mortality. Hence, NF-kB seems to be a promising target, and inhibitors that can modulate the abnormal functioning of NF-kB have a vital role in controlling disease progression and avoiding mortality. Thus, the selected five targets have key roles in different stages of pathogenesis and immune response. The phytochemicals that can simultaneously inhibit the activity of all these targets can be recommended as a lead for further investigation leading to the discovery of safe, effective, and stable drugs against rapidly mutating pathogens such as SARS-CoV-2. This multitargeting drug discovery approach is now getting more acceptance [11]. The docked results revealed that out of 97 phytochemicals derived from G. gummi-gutta, 18 of them have an inhibitory effect on all five selected targets, which indicates the significant anti-SARS-CoV-2 activity of the selected plant. To optimise the best lead, the top five lead molecules were subjected to protein-ligand interactions and ADMET property analysis and the results suggested that the compound amentoflavone showed good docking scores and protein-ligand interactions along with favourable ADMET properties against the selected targets compared to the reference drug, remdesivir. The compound establishes H-bond and hydrophobic interactions with Gln493, Gly502, and Tyr505 amino acid residues, which are the interactive residues of SP RBD with the human ACE2 receptor [18]. In addition, the lead compound interacts with Arg514 of ACE2, which has been identified as a crucial residue in substrate selectivity, and also establishes interaction with His41 and Cys145, the catalytic dyad of Mpro, one of the most highly conserved enzymes among the coronaviruses [34,35]. Therefore, the compound amentoflavone possesses significant stable binding interactions with multiple targets of SARS-CoV-2. This was substantiated by MD simulation, where the lead compound was compared with the inhibitor bound to Mpro. The parameters analyzed for 100 ns of simulations were RMSD, RMSF, the radius of gyration, and hydrogen bonding, and the results revealed that amentoflavone showed better stability than natural inhibitor-bound Mpro.
The compound amentoflavone, a biflavonoid of apigenin, possesses wide pharmacological activities, including anti-viral, anti-fungal, anti-inflammatory, antioxidant, and anti-cancer [36]. In addition, various studies have suggested the anti-viral properties of amentoflavone against respiratory syncytial viruses, herpes simplex virus, influenza virus [37], human immunodeficiency virus [38], Coxsackievirus B3 [39], dengue virus [40], and hepatitis virus [41]. Recently, molecular docking studies have predicted the potential of amentoflavone to inhibit SAR-CoV-2 via binding to Mpro [42]. The current study provides insight into whether amentoflavone is an ideal anti-COVID-19 drug candidate since it shows multiple target inhibitions, such as 1) entry inhibition, 2) inhibition of viral replication, 3) inhibition of viral multiplication, and 4) protection of host cells from inflammatory responses. It is conceivable that, based on the current in silico results, amentoflavone can be considered a potential drug candidate against SARS-CoV-2. However, further in vitro and in vivo studies are to be recommended for practical confirmation.
CONCLUSION
In silico screening of 97 phytomolecules from G. gummi-gutta against five targets, each with a key function in different stages of viral infection and replication revealed that 18 phytochemicals have inhibitory activity on all targets. Based on binding energy, potency, and ADMET analysis, the compound amentoflavone was identified as the best lead against SARS-CoV-2. The results confirm the traditional use of the plant against viral diseases. Further experimental studies are necessary to match the in silico results.
ACKNOWLEDGMENT
The authors are thankful for the facilities and support extended by the Director, KSCSTE-Jawaharlal Nehru Tropical Botanic Garden and Research Institute, and for the research fellowship awarded to the first author by the Council of Scientific and Industrial Research (CSIR), Govt. of India.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
Financial support by Kerala State Council for Science Technology and Environment, Govt. of Kerala and CSIR, Govt. of India.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. covid19.who.int [Internet]. WHO COVID-19 dashboard. Geneva, Switzerland: World Health Organization; 2020 [cited 2023 Sep 19]. Available from: https://covid19.who.int/
2. Gil C, Ginex T, Maestro I, Nozal V, Barrado-Gil L, Cuesta-Geijo MÁ, et al. COVID-19: drug targets and potential treatments. J Med Chem. 2020;63(21):12359–86. CrossRef
3. Janik E, Niemcewicz M, Podogrocki M, Saluk-Bijak J, Bijak M. Existing drugs considered as promising in COVID-19 therapy. Int J Mol Sci. 2021;22(11):5434. CrossRef
4. Naidoo D, Roy A, Kar P, Mutanda T, Anandraj A. Cyanobacterial metabolites as promising drug lead against the Mpro and PLpro of SARS-CoV-2: an in-silico analysis. J Biomol Struct Dyn. 2021;39(16):6218–30. CrossRef
5. Jiang Y, Yin W, Xu HE. RNA-dependent RNA polymerase: structure, mechanism, and drug discovery for COVID-19. Biochem Biophys Res Commun. 2021;538:47–53. doi: https://doi.org/10.1016/j.bbrc.2020.08.116
6. Singh S, Sk MF, Sonawane A, Kar P, Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J Biomol Struct Dyn. 2021;39(16):6249–64. CrossRef
7. Keerthi Sugathan J, Sreekumar S, Biju CK. Identification of lead molecules against multi-target of SARS-CoV-2 from Carica papaya L. through in-silico method. Int J Pharm Sci Drug Res. 2023;15(2):124–31. CrossRef
8. Rodriguez-Garcia A, Hosseini S, Martinez-Chapa SO, Cordell GA. Multi-target activities of selected alkaloids and terpenoids. Mini Org Chem. 2017;14(4):272–9. CrossRef
9. Kabir A, Muth A. Polypharmacology: the science of multi-targeting molecules. Pharmacol Res. 2022;176:106055. CrossRef
10. Anilkumar AT, Manoharan S, Balasubramanian S, Perumal E. Garcinia gummi-gutta: phytochemicals and pharmacological applications. BioFactors. 2023;49(3):584–99. CrossRef
11. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–91. CrossRef
12. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx: methods. Mol Biol. 2015;1263:243–50. CrossRef
13. Trott O, Olson AJ, AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. CrossRef
14. Schrödinger L, DeLano W. PyMOL [Internet]; 2020. Available from: http://www.pymol.org/pymol
15. discover.3ds.com [Internet]. San Diego, CA: BIOVIA, Dassault Systèmes; 2020 [cited 2023 Sep 19]. Available from: https://discover.3ds.com/
16. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci rep. 2017;7(1):42717. CrossRef
17. Pires DEV, Blundell TL, Ascher DB. pkCSM predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–72. CrossRef
18. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACR2 receptor. Nature. 2020;581:215–20. CrossRef
19. Choubey A, Dehury B, Kumar S, Medhi B, Mondal P. Naltrexone a potential therapeutic candidate for COVID-19. J Biomol Struct Dyn. 2022;40(3):963–70. CrossRef
20. Sarah H, Kummetha VIR, Tiwari VSK, Huante MB, Clark AE, Wang S et al. Discovery and mechanism of SARS-CoV-2 main protease inhibitors. J Med Chem. 2022;65:2866–79. CrossRef
21. Hu Q, Xiong Y, Zhu HH, Zhang YN, Zhang YW, Huang P, et al. The SARS-CoV-2 main protease (Mpro): structure, function, and emerging therapies for COVID-19. MedComm. 2022;3(e151):1–27. CrossRef
22. Aftab SO, Ghouri MZ, Masood MU, Haider Z, Khan Z, Ahmad A, et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J Transl Med. 2020;18:275. CrossRef
23. Su CM, Wang L, Yoo D. Activation of NF-kB and induction of proinflammatory cytokine expressions mediated by ORF7 a protein of SARS-CoV-2. Sci Rep. 2021;11:1364. CrossRef
24. Kircheis R, Haasbach E, Lueftenegger D, Heyken WT, Ocker M, Planz O. NF-kB pathway as a potential target for treatment of critical stage COVID-19 patients. Front Immunol. 2020;11:598444. CrossRef
25. Choudhary S, Singh PK, Verma H, Singh H, Silakari O. Success stories of natural product-based hybrid molecules for multi-factorial diseases. Eur J Med Chem. 2018;151:62–97. doi: https://doi.org/10.1016/j.ejmech.2018.03.057
26. Keerthi Sugathan J, Sreekumar S, Biju CK. In silico validation of clove as a potential nutraceutical against SARS-CoV-2. Int J Pharm Sci Res. 2022;13(11):4544–53. CrossRef
27. Deepa V, Sreekumar S, Biju CK. In-silico validation of anti-russell’s viper venom activity in Phyllanthus emblica L. and Tamarindus indica L. Int J Pharm Sci Drug Res. 2018;10(4):217–26. CrossRef
28. Veeresham C. Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res. 2012;3(4):200–1. CrossRef
29. Shityakov S, Förster C. In silico predictive model to determine vector-mediated transport properties for the blood–brain barrier choline transporter. Adv Appl Bioinforma Chem. 2014;2:23–36. CrossRef
30. Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129–43. CrossRef
31. Ramírez D, Caballero J. Is it reliable to take the molecular docking top-scoring position as the best solution without considering available structural data? Molecules. 2018;23(5):1038. CrossRef
32. Wrobel AG. Mechanism and evolution of human ACE2 binding by SARS-CoV-2 spike. Curr Opin Struct Biol. 2023;81:102619. CrossRef
33. Pang X, Xu W, Liu Y, Li H, Chen L. The research progress of SARS-CoV-2 main protease inhibitors from 2020 to 2022. Eur J Med Chem. 2023;22:115491. CrossRef
34. Rushworth CA, Guy JL, Turner AJ. Residues affecting the chloride regulation and substrate selectivity of the angiotensin-converting enzymes (ACE and ACE2) identified by site-directed mutagenesis. FEBS J. 2008;275(23):6033–42. CrossRef
35. Rocha RE, Chaves EJ, Fischer PH, Costa LS, Grillo IB, Da Cruz LE, et al. A higher flexibility at the SARS-CoV-2 main protease active site compared to SARS-CoV and its potentialities for new inhibitor virtual screening targeting multi-conformers. J Biomol Struct Dyn. 2002;40(19):9214–34. CrossRef
36. Yu S, Yan H, Zhang L, Shan M, Chen P, Ding A, et al. A review on the phytochemistry, pharmacology, and pharmacokinetics of amentoflavone, a naturally-occurring biflavonoid. Molecules. 2017;22(2):299. CrossRef
37. Ma SC, But PP, Ooi VE, He YH, Lee SH, Lee SF, et al. Antiviral amentoflavone from Selaginella sinensis. Biol Pharm Bull. 2001;24:311–2. CrossRef
38. Lin YM, Anderson H, Flavin MT, Pai YH, Mata-Greenwood E, Pengsuparp T, et al. In vitro anti-HIV activity of biflavonoids isolated from Rhus succedanea and Garcinia multiflora. J Nat Prod. 1997;60:884–8. CrossRef
39. Wilsky S, Sobotta K, Wiesener N, Pilas J, Althof N, Munder T, et al. Inhibition of fatty acid synthase by amentoflavone reduces coxsackievirus B3 replication. Arch Virol. 2012;157:259–69. CrossRef
40. Coulerie P, Nour M, Maciuk A, Eydoux C, Guillemot JC, Lebouvier N, et al. Structure-activity relationship study of biflavonoids on the dengue virus polymerase DENV-NS5 RdRp. Planta Med. 2013;79:1313–8. CrossRef
41. Lee WP, Lan KL, Liao SX, Huang YH, Hou MC, Lan KH. Inhibitory effects of amentoflavone and orobol on daclatasvir-induced resistance-associated variants of hepatitis C virus. Am J Chin Med. 2018;46:835–52. CrossRef
42. Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–88. CrossRef
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website link: [https://japsonline.com/admin/php/uploadss/4274_pdf.pdf]