
INTRODUCTION
Amoxicillin, a penicillin-derivative antibiotic, is one of the drugs that plays a vital role in overcoming bacterial infection. The synthesis of amoxicillin is possible through the production of the precursor, 6-aminopenicillanic acid (6-APA), from the catalysis reaction of an enzyme called penicillin-G acylase (PGA). This enzyme catalyzes the hydrolysis of penicillin such as penicillin-G by releasing the acyl group to generate 6-APA and side product phenylacetic acid (PAA) [1]. On top of that, PGA could not only hydrolyze β–lactam antibiotics, but also able to synthesize the other penicillin derivatives thus forming semisynthetic antibiotics [2]. The wide application and great potential of PGA in catalyzing hydrolysis and synthesis of various substrates and semisynthetic antibiotics, respectively, make the development of PGA is essential.
Chemical production of antibiotics in the past has been replaced by a safer and more environmentally friendly method, which is through the production of recombinant protein by microorganisms as the host expression system. Several researches have been carried out through various phases to develop the potential of PGA including recombination, gene expression, enzyme isolation and purification, and enzyme activity tests by different recombinant hosts. The cloning and production of recombinant PGA from Escherichia coli and Bacillus megaterium through host cells E. coli BL21(DE3) and DH5α with many optimizations were reported. The gene expression and isolation of PGA were satisfactory, but the activity of the enzyme in enzymatic reaction is considered low and not optimal yet [3]. The low enzyme activity of PGA is likely influenced by the weak binding of the enzyme and substrate. This hypothesis led us to a further approach to increase the enzyme activity, which is by improving the binding strength between enzyme and substrate.
As mentioned above, one of the factors that influence the enzyme activity is the binding of the enzyme and its substrate where the optimal binding of them could potentially improve the enzyme activity. PGA’s active site consists of catalytic and substrate-binding domains with specific residues function in each domain. Its substrate-binding domain consists of subdomain 1 (S1) which binds to the acyl group of substrate and subdomain 2 (S2) which interacts with nucleophiles [4]. Many researchers tried to mutate the residues in the active site through site-directed mutagenesis and many mutants were found to be able to improve the enzyme activity and prove the role of the residues in enzymatic reaction [5,6].
The earlier screening through an in silico approach could serve as an effective way in terms of time, energy, and expense consumption to predict the potential of mutants before conducting the laboratory research. This research intended to find potential PGA mutants by computationally mutating some amino acid residues in the S1 domain that have not yet been widely discussed or mutated. Residues βPro22, βSer67, βThr68, βTrp154, βLeu253, and βPhe256 were substituted by other six residues to create 36 PGA mutants with single amino acid substitution, and the effect of mutation on the enzyme-substrate binding was studied.
The purpose of this research was to study the interaction of PGA mutants with penicillin-G and select the potential mutant based on the affinity strength which will be investigated deeper. The effect of site-directed mutagenesis was studied by conducting the transformation of recombinant plasmid, gene expression with different inducers, protein isolation from different cell fractions, and enzymatic activity.
MATERIALS AND METHODS
Homology modelling
The amino acid sequence of α and β subunits were extracted from the PGA amino acid sequence originating from E. coli in NCBI with accession number AAA24324.1 [7]. This sequence serves as the control model and template for mutations. The sequence for mutants was generated by BioEdit [8] by replacing target residues with other residues displayed in Table 1. All protein models were built based on PGA model 1K7D [9] as the template. The α and β subunits of each mutant were built separately in SWISS-MODEL [10] before being combined into a complex by PyMOL [11].
Protein and ligand preparation
Each united protein model was prepared by AutoDockTools 4.2 [12] including the addition of polar hydrogen, a merge of nonpolar hydrogen, the addition of Kollman charge, and the assignment of AD4 type atom. Ligand was prepared by extracting penicillin-G from crystal structure 1FXV [13] by PyMOL and minimizing the energy by PyRx [14]. The preparations included the addition of polar hydrogen, the addition of Gasteiger charge, the assignment of AD4 type atom, and the setting of rotatable bond (five bonds) and active torsion (four torsions).
Molecular docking
Docking validation was performed by AutoDockTools 4.2 with crystal structure 1FXV and by a similar protocol, molecular docking for all PGA mutants was proceeded. To conduct the molecular docking, a grid parameter file (GPF) was first prepared with grid box dimensions 80 × 80 × 80, spacing 0.375 Å, and a grid box centered on the ligand. Then, the docking parameter file (DPF) was also prepared with the rigid file of the macromolecule, genetic algorithm as the search parameter and Lamarckian genetic algorithm as output. Using these GPF and DPF, molecular docking was conducted and the result in the form of binding affinity can be observed from the output file. The conformation from the lowest binding affinity that indicates the best binding energy was further compared between each mutant [15].
Transformation of mutant recombinant plasmid
This research used the wild-type PGA gene optimized in previous research [16] by GenScript through its OptimumGene™ codon optimization analysis algorithm. The gene codon was optimized based on the codon usage bias and many other optimization parameters for expression in E. coli. Using the optimized gene sequence, the mutant gene pgaEcβThr68Tyr was created (Supplementary Fig. S1) and constructed in pET-22b(+) plasmid by GenScript. The recombinant plasmid was then transformed into E. coli BL21(DE3) by 42°C and 45 seconds of heat shock.
Escherichia coli BL21(DE3) was first made competent by MgCl2–CaCl2 0.1 M and CaCl2 0.1 M [16]. Then, the amount of 100 μl of transformant culture was spread on an LB plate containing 50 μg/ml ampicillin. The plate was then incubated at 37°C overnight to form bacterial colonies. Colony polymerase chain reaction (PCR) was carried out by T7 Promoter and pgaEc specific (5’-GTA CCA ACA AAG ATC ATC G-3’) primer as forward and reverse primers, respectively. Each PCR reaction was conducted in a final volume of 20 μl, containing 100 μM of dNTPs, 2.5 u of DNA Taq polymerase, and 2 mM MgCl2 obtained from the ready-mixed Promega’s Gotaq Green Master Mix. The PCR reaction also contains 0.4 μM forward primer, 0.4 μM reverse primer, 100 ng of DNA template, and finally nuclease-free water to add up to the final volume of the reaction mixture. The PCR program included a pre-denaturation step at 95°C for 4 minutes followed by 27 cycles of denaturation at 95°C for 30 seconds, annealing at 47°C for 30 seconds, and extension at 72°C for 1 minute before the final extension at 72°C for 5 minutes. Amplicons were observed on 1% of gel agarose electrophoresis.
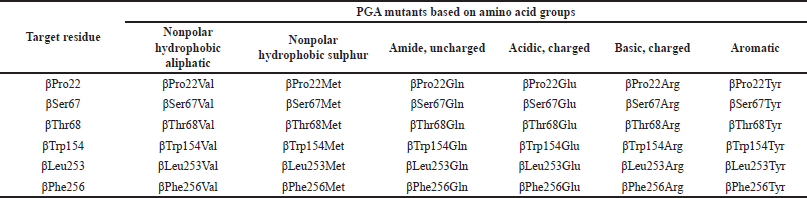 | Table 1. 36 PGA mutants generated from the substitution of 6 target active site residues into 6 residues representing each amino acid group. [Click here to view] |
Isolation of mutant recombinant plasmid
Plasmid isolation from transformant culture was implemented by FavorPrep™ Plasmid Extraction Mini Kit. By forward T7 Promoter and reverse T7 Terminator primers, PCR was performed with the plasmid isolate as the DNA template, using the same PCR condition stated in the section “Transformation of mutant recombinant plasmid,” which was followed by the separation of PCR products through 1% of gel agarose electrophoresis.
Expression of mutant recombinant PGA
A volume of 1 ml of starter culture of transformant was inoculated to 50 ml LB broth containing 50 μg/ml of ampicillin. Next, the bacterial culture was incubated and shaken at 170 rpm and 37°C to obtain the OD600 of 0.5–0.7. After reaching the OD value, 0.05 mM IPTG [3] and 1.5% arabinose [16] were added to different culture replicates. Then, incubation was continued in a shaker incubator at 150 rpm and 37°C for 17 hours.
Recovery of mutant and wild-type recombinant PGA
After gene expression, the transformant culture was centrifuged at 3,500 rpm and 4°C for 30 minutes. The pellet was resuspended with 5 ml of 0.1 M potassium phosphate pH 7.8 buffer before centrifuging again for 15 minutes. Resuspension of the pellet was repeated once and sonication was performed on the bacterial suspension for 10 cycles with 30 seconds of start and stop duration each. The sonicated suspension was then separated through 5,000 rpm of centrifugation for 20 minutes at 4°C. The supernatant and pellet were recovered as S1 and P1 fractions, respectively. Then, 3 ml of N-laurylsarcosine solubilization buffer (5% n-propanol, 5% dimethyl sulfoxide, and 0.2% N-laurylsarcosine) was added and the suspension was incubated at 4°C for 90 minutes. After solubilization, the suspension was separated through 5,000 rpm of centrifugation for 20 minutes. The pellet (P2) was resuspended by buffer and the supernatant was filtered through the 15 ml Amicon® ultra centrifugal filters (MWCO 10,000) before the filtrate (S3) was obtained. The concentration of total protein was measured from all cell fractions by bovine serum albumin as standard. In addition, the uniform concentration of these protein fractions was prepared by an SDS sample buffer before the SDS-PAGE running in 12% resolving and 5% stacking polyacrylamide gel.
Determination of enzymatic activity of mutant and wild-type PGA
An amount of 1 ml of protein fractions S1 and S3 with similar concentration (1 mg/ml) was reacted with 1 ml 4.5% K penicillin-G in 0.1 M potassium phosphate pH 7.8 buffer for 30 minutes in a shaker incubator at 37°C of temperature. After the enzymatic reaction, 1 ml of 99% acetonitrile and 1% acetic acid were added to stop the enzyme reaction followed by centrifugation for 2 minutes at 4,500 rpm. The amount of 1 ml of supernatant was moved to a test tube that contained 3 ml of para-dimethylaminobenzaldehyde (p-DAB) reagent (0.5% p-DAB, 2% of 20% acetic acid, 1% of 0.05 M NaOH). After leaving the mixture for 4 minutes at room temperature, the solution was measured by a spectrophotometer at 415 nm of wavelength.
RESULT AND DISCUSSION
Homology modeling
All of the constructed mutant PGA models have global model quality estimate values ranging from 0.93 to 0.96. The score represents the quality of the models built when compared to the template model and the score approaching 1 indicates a good model [10,17]. The QMEANDisCo global scores of all models that evaluate the atomic distance consistency between the target model and ensembled experimental models are close to 1, which is between 0.82 ± 0.06 and 0.88 ± 0.05 which also indicates good models [10,18]. The quality of the protein models is also supported by the QMEAN Z-score which is located in the <1 area, indicating the closeness of the models with other experimental crystal proteins.
Docking validation
Docking validation was performed to ensure the validity of the docking protocol before the docking of the constructed models. This step includes the re-docking of the native ligand with its native protein from the experimental structure. The deviation of the result then could be assessed based on the root mean square deviation (RMSD) value [19]. The crystallized structure of PGA, that is 1FXV, was used in this validation because it is the crystallized PGA model that existed with a similar ligand intended for this research. The re-docking of the complex resulted in ten ligand conformations but the lowest RMSD value was chosen to be considered regardless of the energy rank [20].
Among all of the docked conformations, the lowest RMSD (1.29) was shown by conformation 3 with a binding affinity of −4.99 kcal/mol. The RMSD value is used to compare the pose between the docking result and the existing crystal structure. The value of 1.5 Å as the maximum RMSD value for a docking method is recommended [20]. However, this value which is set for the ligand with a low number of rotatable bonds is 2. However, the ligand in this study is penicillin-G which is highly rotatable with five rotatable bonds and four active torsions which makes this RMSD value more than sufficient to be validated. It was also stated that 1.5–2 Å is acceptable depending on the ligand. Therefore, the docking protocol used in this research was validated, proven by the low deviation of docked conformation from the experimental structure (Fig. 1).
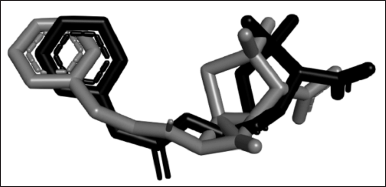 | Figure 1. The superimposition of penicillin-G from the re-docked structure (grey) and crystal structure 1FXV (black). The structure of the re-docked penicillin-G deviated from its experimental crystal structure in RMSD 1.29, which is an acceptable value for docking to be considered validated. [Click here to view] |
Molecular docking
The lowest binding affinity from all models varied, ranging from −6.46 to −4.27 kcal/mol (Fig. 2). When compared to the wild type as a control model, only six mutant models resulted in the lower or better binding affinity than control (βPhe256Met, βPhe256Arg, βPhe256Tyr, βLeu253Met, βThr68Arg, and βThr68Tyr) whereas the rest 30 models had higher or lower binding affinity than control. The variety of values was expected as the change in even a single amino acid could cause a change in the protein structure and lead to different modes of interaction between the protein and its ligand.
The target residues were substituted into other six residues that represent six groups of amino acids as stated in the methods (Table 1). The categorization was based on the standardized classes of amino acids from Immunogenetics (IMGT), the international IMGT information system useful for mutation and protein engineering purposes (https://www.imgt.org). It would be more time-consuming and less effective if all 19 amino acids were used to replace the target amino acid. These amino acids (Val, Met, Gln, Glu, Arg, and Tyr) were chosen from each group because these amino acids are structurally the largest amino acids that are predicted to lead to a significant change in protein structure and interaction.
There was no uniform pattern that represents how the size of amino acid could influence the binding between enzyme and substrate from previous studies. The mutation of αMet142 and αPhe146 into alanine has been known to reduce the hydrolysis of PGA on penicillin-G [21]. Based on the IMGT database, methionine, phenylalanine, and alanine are categorized as large, very large, and very small residues, respectively, which indicates the high difference in the size of amino acids. In addition, the mutation of αArg145, a large amino acid into lysine which is also a large amino acid results in significant results where the hydrolysis of penicillin-G is lower than wild type [22]. The change of αPhe146 into tyrosine, which is in the same size category as αPhe146, leads to the increase of affinity between PGA and phenylglycinamide in ampicillin synthesis [23]. However, to maximize the change of structure in the protein and potentially generate significant observation, the biggest amino acid was considered to be used in this study [24].
 | Figure 2. Comparison of binding affinity among PGA mutants and with wild-type PGA. Out of 36 PGA mutants, 6 of them (βPhe256Met, βPhe256Arg, βPhe256Tyr, βLeu253Met, βThr68Arg, and βThr68Tyr) generate binding affinity that is lower than that of the control (wild-type PGA) when binding to the penicillin-G. [Click here to view] |
Each molecular docking result of six mutants showed that penicillin-G interacts directly with various residues in the active site of the PGA model, leading to the alteration of structure (Table 2). The control model from wild-type PGA interacts with seven residues. Theoretically, although there are many important residues in active sites of PGA, residues that have function catalytically in enzyme reactions are Ser1, Gln23, Asn241, and Ala69. The nucleophile of the hydroxyl group of Ser1 attack C carbonyl on the amide bond of penicillin-G. The Gln23 indirectly interacts with the substrate to help enhance the nucleophilicity of Ser1. Oxy-anion tetrahedral intermediate will be formed and then will be stabilized by Asn241 and Ala69 by a hydrogen bond. Then, an acyl-enzyme complex will be produced and 6-APA will be released from the active site before deacylation takes place and PAA and enzyme are released [25].
The interaction in the control model with the ligand displays interaction with all residues needed except for Asn241. The βAsn241 residue plays a crucial role in not only balancing the negative charge in the tetrahedral reaction intermediate but also helping in determining the position of ligand and active site Ser1 [26]. This residue together with Ala69 binds with O16 of penicillin-G (Fig. 3) after the formation of an intermediate state after a Ser1 nucleophilic attack. The interaction of control shows that Ser1 interacts with O8 instead of O16. Based on the catalytic mechanism mentioned, there is a possibility that the interaction conformation captured in the docking consists of the step before the nucleophilic attack, which explains the absence of a reaction of βAsn241 with the O16 that binds to C15. The crystal structure of PGA, 1FXV also shows no interaction with βAsn241. However, when the interaction visualization is expanded, βAsn241 is seen to interact indirectly with ligand where it interacts with Ser1. Based on the presence of direct interaction with catalytic residues Ser1, Ala69, and βAsn241 and indirect interaction with Gln23 through hydrogen bond with Ser1, mutant βPhe256Arg, βPhe256Tyr, βLeu253Met, and βThr68Tyr are catalytically possible.
Other than the residues that function catalytically, many other residues in active sites function in the substrate-binding domain. Residues αMet142, αPhe146, βPhe24, βPhe57, βTrp154, βIle177, βSer67, βPro22, βGln23, βVal56, βThr68, βPhe71, βLeu253, and βPhe256 in S1 subdomain plays a role in interaction of hydrophobic pocket with substrate’s acyl group. In addition, residues αArg145, αPhe146, βPhe71, and βArg263 in the S2 subdomain are important in the binding of the nucleophilic group [4]. All of the ligands in PGA models interact with most of the residues mentioned, mostly through a hydrophobic bond with the acyl part of the substrate and a pi-sulfur bond with sulfur on the thiazolidine ring of penicillin-G. There is an exception for Ser386 in the wild-type model and Ser386 and Asn388 in βThr68Tyr models because these residues have never been determined as active sites. A study that mutates αAsp148, a site that is not known as an active site in penicillin acylase discovered its important function in transitions in conformational changes because of its position in the catalytic loop of the protein [4]. Based on the position of these Ser386 and Asn388 residues which are on the surface of the enzyme’s inner side, these residues are in direct contact with the substrate and possibly play an important role in the conformational change of protein and binding of the substrate.
The mutant PGA with the lowest binding affinity, βThr68Tyr, has three potential conformations with binding affinity −6.46, −6.29, and −5.65 kcal/mol. It indicates the strongest binding affinity among other mutants. However, when considering the number of residues interacted, this mutant does not interact with the highest number of residues (Table 2). However, the number of residues does not mainly determine the strength of enzyme-substrate binding, but it is determined by the binding mode in which the molecule interacts with the ligand. An ideal scoring function will result in the lowest binding energy and at the same time a similar binding mode as in crystal structure but for a precaution, binding mode should be considered more than the binding affinity [27].
The βThr68Tyr mutant results in the best binding affinity which indicates a stronger bond between enzyme and substrate during enzymatic reaction. Moreover, the three available conformations from this mutant represent more favorable poses when the ligand interacts with PGA. This mutant also shows interaction with residues for binding of ligand to be possible and catalysis reaction to occur. Nevertheless, the mutants βPhe256Tyr, βPhe256Arg, and βLeu253Met should not be ignored since these mutants also interact with ligands through catalytic and substrate-binding residues other than the fact that these mutants have better binding affinity than the control does. However, due to the relatively slight difference in their binding affinity compared to that of in control, there is less possibility that the enzyme activity would change significantly.
Transformation of mutant recombinant plasmid
The transformation of mutant recombinant plasmid pET22b-pgaEcβThr68Tyr into competent E. coli BL21(DE3) results in the formation of bacterial colonies on the LB agar plate (Fig. 4). The principle of transformation still applies to the mutant plasmid used in our research where the application of divalent Ca²+ could reduce the repulsion between bacterial cell and plasmid by neutralizing the charge hence facilitating the plasmid to be absorbed by the cell [28]. In addition, the additional divalent such as Mg²+ that was applied to generate competent cells could increase the efficiency of transformation just like what has been reported [29]. It explains the plentiful transformant colonies formed on the agar plate. Nevertheless, single colonies were still able to be distinguished and isolated from the agar media.
The colonies were confirmed to contain the mutant PGA gene, indicated by the appearance of DNA bands on electrophoresis gel after a colony PCR. As represented by Figure 5, DNA bands appeared within the targeted band size which is 537 bp from all colonies (A, B, and C) that were used as PCR templates. The appearance of these bands is similar to the size of the DNA band from positive control (+), which indicates the ability of the transformation method applied to transform the mutant recombinant plasmid and the cell host to grow on media while carrying the plasmid.
The bacterial colonies were also assured to carry the mutant plasmid pET22b-pgaEcβThr68Tyr, based on the appearance of PCR product after the plasmid PCR of plasmid isolates (Fig. 6). The amplicons possess the targeted size of the whole PGA gene (2,538 bp). These amplicon bands do not appear from the negative control, plasmid isolates of E. coli BL21(DE3), as the cells in the mentioned culture do not contain recombinant plasmid.
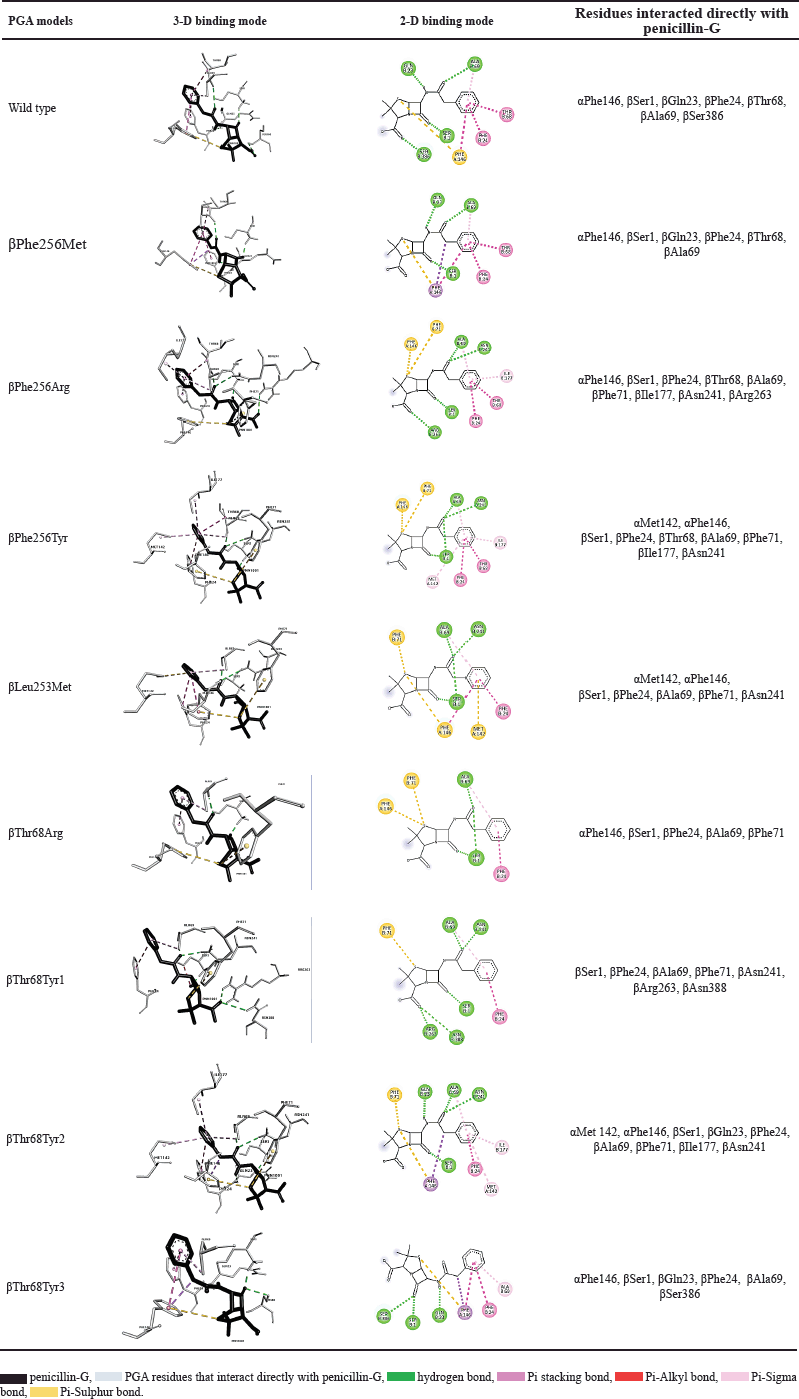 | Table 2. The conformation and interaction of PGA models and penicillin-G. [Click here to view] |
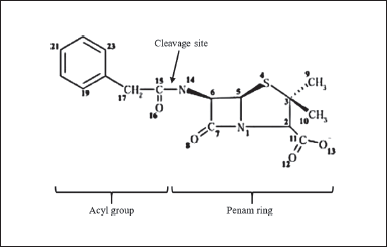 | Figure 3. Structure of penicillin-G consists of an acyl group and a penam ring. The PGA enzyme will cleave the substrate at the cleavage site of the amide group and then release the acyl group (PAA) and penam ring (6-APA) [25] . [Click here to view] |
 | Figure 4. Colonies of E. coli BL21(DE3)-pET22b-pgaEcβThr68Tyr formed on the LB agar plate with 50 μg/ml ampicillin after a spread plate of 100 μl transformant culture and an overnight incubation at 37°C. [Click here to view] |
Expression and recovery of mutant and wild-type recombinant PGA
Escherichia coli BL21(DE3) as the expression host of the mutant and wild-type recombinant PGA were fermented and resulted in the noticeable protein yield regardless of the level differences in each culture. It indicates a good expression of both mutant and wild-type PGA by the cultivation condition, optimized codon, and expression host in this research. The gene expression process generated different levels of total protein in each cell fraction (Fig. 7). The total protein concentration in the insoluble fraction (P1) from all cultures, ranging from 27.97 to 30.26 mg/ml, is obviously higher than the concentration in their respective soluble fractions (S1) which are between 0.95 and 1.97 mg/ml. The P1 fractions represent insoluble fraction gained after cell lysis, containing the cell debris from membranes and aggregated form of protein whereas S1 fractions depict the cytoplasmic fraction that contains soluble protein [30]. In this research, the S1 fraction also contains soluble proteins from cell periplasmic space where mature PGA is present.
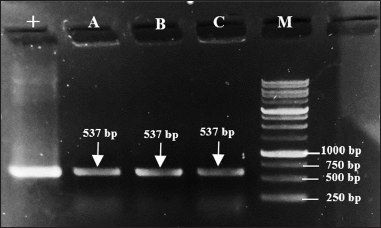 | Figure 5. Target DNA bands (537 bp) formed on 1% electrophoresis gel after colony PCR. Lane 1 (+): pure mutant plasmid pET22b-pgaEcβThr68Tyr, lane 2 (A): colony A, lane 3 (B): colony B, lane 4 (C): colony C, and lane 5 (M): 1 kb DNA ladder. [Click here to view] |
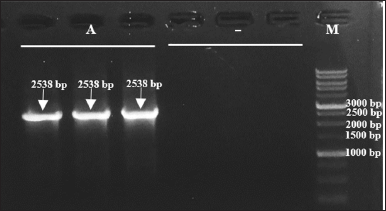 | Figure 6. Target DNA bands (2,538 bp) formed on 1% electrophoresis gel after plasmid PCR. Lanes 1–3 (A): plasmid isolates from transformant E. coli BL21(DE3)-pET22b-pgaEcβThr68Tyr, lanes 4–6 (−): plasmid isolates from E. coli BL21(DE3), and lane 7 (M): 1 kb DNA ladder. [Click here to view] |
Although the total protein concentration values do not specifically indicate the concentration of PGA, the visualization of protein bands after separation on SDS-PAGE gel could help to determine the quality and presence of our protein target from each fraction. The different value of protein concentration in P1 and S1 fractions is confirmed by the presence of the thicker bands of P1 on SDS-PAGE gel when compared to the S1 bands (Figs. 8 and 9). Arabinose inducer expressed more β subunits but fewer inclusion bodies in P1 when compared to S1 (Fig. 8). This might suggest that the absence or low formation of inclusion bodies during the protein expression was induced by arabinose. In contrast, SDS-PAGE of the IPTG-induced culture visualizes the similar quality of the β subunit band from P1 and S1 but thicker inclusion bodies in P1. This observation shows the higher amount of inclusion bodies caused by IPTG, especially when expressing βThr68Tyr mutant results in a noticeably thicker band of inclusion bodies (Fig. 9). We assume that the higher concentration of proteins in P1 from arabinose induction was contributed by PGA proteins while that of from IPTG induction was contributed mainly by the formation of inclusion bodies. The α subunit band of PGA was not observable in both gels (Figs. 8 and 9) which is probably due to its low concentration.
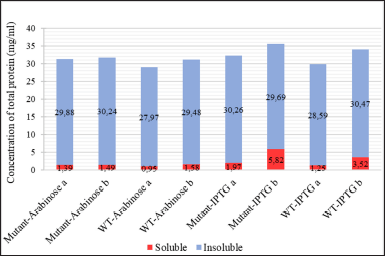 | Figure 7. The concentration of total protein in cell fractions after sonication (a: S1 and P1) and after solubilization treatment with N-laurylsarcosine (b: S2 and P2) from different cell cultures: mutant PGA βThr68Tyr expression induced by arabinose (mutant-Arabinose), wild-type PGA expression induced by arabinose (WT-Arabinose), mutant PGA βThr68Tyr expression induced by IPTG (mutant-IPTG), wild-type PGA expression induced by IPTG (WT-IPTG).: soluble fraction in the form of supernatant, : insoluble fraction in the form of pellet. [Click here to view] |
The higher protein concentration in P1 indicates the high formation of aggregated proteins with higher density, hence, settled in the base of the microtube after centrifugation. This is a common occurrence in protein overexpressions due to the incomplete folding of protein where the formation of inclusion bodies is expected [31]. Other than because of the use of strong promoters such as T7 that boosts the heterogenous expression, the application of optimized codon and E. coli as expression hosts could lead to the imperfect folding of proteins that will aggregate to form inclusion bodies [32]. It is also possible that the cell host could not withstand the high expression stress when fulfilling the metabolic prerequisite, especially after the translation where proteins are attempting to be modified through folding [33].
Our study used a codon-optimized gene along with a T7 promoter provided by the construction of pET22b-pgaEc recombinant plasmid that was transformed and expressed in E. coli, which led to the formation of inclusion bodies. However, this approach helps better the expression of synthetic PGA genes in the cell host after many optimizations [3,16]. We had anticipated the formation of aggregates hence we increased the number of cycles and duration during sonication. The 10 cycles with 30 seconds of sonication in each cycle have increased the soluble total protein yield compared to the previous reports in the same series of research [16]. It was also revealed that cell sonication could physically affect the structure of aggregates by breaking them into the desired size favored for better dissolution [34]. Furthermore, we attempted to apply low temperature (20°C) for induction and fermentation conditions to slow down the expression process so that an optimal modification and folding of proteins could take place after the translation [35].
Because there is a presence of proteins in aggregated form in P1, further steps should be carried out to extract more bioactive proteins from the insoluble fraction. The inclusion bodies are not meaningless as they have their potential as bionanomaterial and even their formation is intentionally induced in protein production [36]. Since the aggregates in inclusion bodies usually consist of a similar type of polypeptides and still possess a secondary structure close to its native, the recovery of the structure is possible through a mild solubilization treatment [37].
The S3 fractions which are the filtered soluble fraction obtained after the solubilization of the insoluble fraction (P1) with N-laurylsarcosine buffer showed an increase in protein level (1.49–5.82 mg/ml) when compared to S1 (Fig. 7). It is in accordance with the thicker visibility of β subunit from S3 compared to S1 in both WT and mutant PGA expression induced by arabinose and IPTG (Figs. 8 and 9). The increase of total protein concentration in the filtrate of solubilized insoluble protein (S3) might be contributed mainly by the increase of protein level after solubilization treatment using N-laurylsarcosine which was able to release the protein from its aggregate form thus solubilizing it. Sarcosyl is a mild detergent that benefits the recovery of protein from the inclusion bodies since the treatment assists the solubilization process better by not denaturing it completely hence the folding process will not be complicated [38]. Then, the removal of denaturant, which in this research was conducted through additional washing and filtration of solubilized insoluble protein, could help refold and generate active protein [39].
The total protein content in P2 fractions (29.48–30.47 mg/ml) which are the solubilization debris mostly show an increase compared to that of P1 (Fig. 7). This phenomenon is due to the increase of β subunit represented by the thicker β subunit and the rise of inclusion bodies indicated by the thicker inclusion bodies in arabinose-induced expression (Fig. 8). It indicates the increase of active protein level, considered from the more visible β subunit of PGA but also the lead to more aggregation showed by the visibility inclusion bodies. Similarly, another research found that solubilization and refolding of inclusion bodies is not an easy task and even though most proteins could be released from their aggregates, most faced the challenge of achieving accurate structure and because of that, the protein could aggregate again [40]. Nevertheless, considering the presence of the β subunit which represents the active form of PGA in the P2 fraction, if further washing and additional centrifugation steps were taken after the solubilization, the active form of protein in P2 would be recovered and might be included in the soluble fraction (S3).
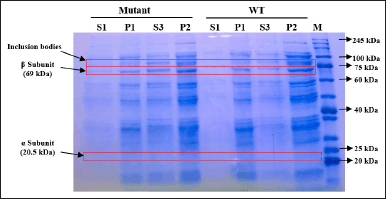 | Figure 8. SDS-PAGE of protein fractions recovered from culture expressed by inducer arabinose. S1: supernatant after cell sonication, P1: pellet after cell sonication, S3: filtrate of solubilized insoluble fraction, P2: solubilization insoluble debris, Mutant: mutant PGA βThr68Tyr, WT: wild-type PGA. [Click here to view] |
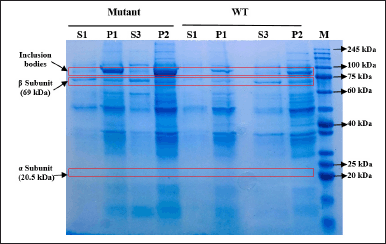 | Figure 9. SDS-PAGE of protein fractions recovered from culture expressed by inducer IPTG. S1: supernatant after cell sonication, P1: pellet after cell sonication, S3: filtrate of solubilized insoluble fraction, P2: solubilization insoluble debris, mutant: mutant PGA βThr68Tyr, WT: wild-type PGA. [Click here to view] |
In IPTG-induced expression, the relative similarity of β subunit from P1 and P2 in WT and mutant PGA indicates the increase of protein concentration is mainly contributed by the rise of inclusion bodies, indicated by thicker P2 bands when compared to P1 (Fig. 9). This observation is consistent with those in arabinose-induced expression where the presence of inclusion bodies and β subunit in solubilization debris (P2) shows the incomplete denaturation or re-aggregation and failure of the released active PGA to complete its accurate structure to be soluble. However in mutant PGA expression, even though the inclusion bodies from P2 are noticeably thicker than P1, the concentration of total protein in P2 does not increase from P1 (Fig. 7). This might relate to the increase of protein content in S3 indicates more proteins from P1 was solubilized from inclusion bodies.
Specific enzyme activity of mutant and wild-type PGA
The enzyme activity of mutant and wild-type PGA was carried out by the soluble fraction represented by the supernatant obtained after sonication (S1) and the filtrate of solubilized insoluble fraction (S3) by reacting the recovered enzyme with its substrate, penicillin-G. The specific enzyme activity of PGA from S1 fraction (1.38–1.93 U/mg) is noticeably higher than that of from S3 fraction (0.45–0.95 U/mg) for both mutant and WT protein expression induced by IPTG (Fig. 10). A similar pattern is also observed from those induced by arabinose where the specific enzyme activity of PGA from S1 fraction (1.73–2.67 U/mg) is higher than that of from S3 fraction (1.7–2.4 U/mg) but the difference is not as remarkable as in IPTG-induced expression.
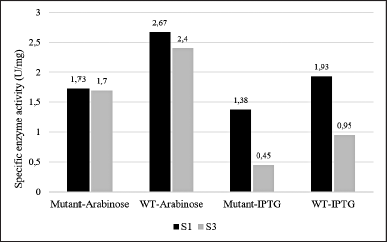 | Figure 10. The specific enzyme activity of PGA from soluble fraction (S1) and solubilized insoluble fraction (S3) from different cell cultures: mutant PGA βThr68Tyr expression induced by arabinose (mutant-Arabinose), wild-type PGA expression induced by arabinose (WT-Arabinose), mutant PGA βThr68Tyr expression induced by IPTG (mutant-IPTG), wild-type PGA expression induced by IPTG (WT-IPTG). Each sample data represents the average of three replicates. The ANOVA analysis displays insignificance (p > 0.05) with p-value = 0.165 between mutant and wild-type PGA and p-value = 0.071 between inducer treatments. [Click here to view] |
The PGA is a hydrolase enzyme that has the ability to break down penicillin-G into 6-APA as the main product and PAA as the side product by cleaving the acyl group on the substrate [1]. The specific enzyme activity values presented in Figure 10 indicate that the PGA obtained from the soluble fraction has a better ability to hydrolyze the enzymatic reaction in comparison to the solubilized insoluble fraction. As we know, an enzyme’s function depends mainly on its structure which is usually developed through processing steps after its translation before it can be mature and functional [41]. PGA is translated in the form of its precursor first before being exported to periplasm. Some subunits including signal peptide, α subunit, spacer peptide, and β subunit made up this precursor that will be modified later. The signal and spacer peptide will then be removed in the periplasm hence generating the bioactive heterodimer of PGA consisting of α and β subunits [42].
The higher protein concentration (Fig. 7) which might represent the higher PGA value and the presence of β subunit (Figs. 8 and 9) which indicates the presence of the active form of PGA in S3 is in contrast with the lower specific enzyme activity when compared to S1 (Fig. 10). This may relate to the implementation of solubilization and refolding steps on insoluble fraction P1. Although more soluble proteins were restored after the solubilization, the released PGA might not have the accurate structure yet due to incomplete refolding. The maturation of penicillin acylases into catalitically active protein is indeed a crucial phase that is usually hindered by either incorrect translocation of PGA precursor, inaccurate folding of PGA in the periplasm, or proteolytic activity of peptidase that is not specific [35]. Therefore, it is important to concurrently apply more than one influential factor as the basis when designing a protein yield improvement [43].
Other than because of the refolding failure of PGA to turn bioactive, the insufficient solubilization or separation of solubilization suspension afterward might also contribute to the low activity of PGA in the S3 fraction. This is assessed from the presence of β subunit in the solubilization debris (P2) that failed to be dissolved in the soluble S3 fraction (Figs. 8 and 9). Using the same solubilizing agent (N-laurylsarcosine), more complicated and further solubilization and refolding were implemented but the solubilized proteins recovered from the inclusion bodies were not as biologically active as the native proteins in the soluble fraction [44]. Since there is a possibility of interference from the solubilizing agent to the protein’s activity, it is suggested to use a lower concentration of N-laurylsarcosine which is 0.05%. Moreover, it is also preferred to avoid the use of high-concentration and strong denaturants such as urea because it would be more challenging to recover the completely denatured protein [45].
The higher difference in the specific enzyme activity value between the S1 and S3 fraction obtained from IPTG-induced expression may have a connection with the higher content of inclusion bodies settled in the insoluble fraction P1 (Fig. 9). Particularly for mutant PGA that shows the most major inclusion bodies demonstrates the lowest specific enzyme activity (0.45 U/mg) from S3 fraction. The formation of more inclusion bodies indicates that less active protein is present in the soluble protein fraction which also could lead to more challenging recovery and refolding after solubilization. On the other hand, the difference in the specific enzyme activity of PGA gained from arabinose-induced expression is not as high and it is associated with the absence or low presence of inclusion bodies in P1 (Fig. 8). Since there is less aggregation formed, more active protein is present in the soluble protein fraction, and the recovery after solubilization would not be as hard. When comparing the specific enzyme activity of PGA expressed by different inducers, those from arabinose induction result in higher enzyme activity than that induced by IPTG in both mutant and wild-type PGA in all parallel fractions. In comparison to IPTG, it was reported that the greater potential of arabinose as an inducer not only produces low inclusion bodies but also assists the processing of the translated precursor PGA into matured PGA [46]. Another study also found that combined induction using IPTG and arabinose could enhance PGA expression and activity when compared to the use of IPTG alone [35].
The comparison between mutant and WT PGA in this study shows higher specific enzyme activity of PGA in wild-type strains from both types of inducer used (Fig. 10). However, the decrease in the values of the PGA activity is proven to be not significant through an unpaired t-test analysis. We speculate that the lower enzyme activity of mutant PGA has something to do with the presence of inclusion bodies in mutant PGA’s S3 fraction induced by arabinose (Fig. 8) and the thicker presence of inclusion bodies in mutant PGA’s P1 and P2 fraction induced by IPTG (Fig. 9). Genetic modification in PGA expression could lead to the accumulation of proteins that are misfolded and not mature enough thus resulting in low stability and activity of the enzyme [47]. Some factors influence protein aggregation, one of them includes the hydrophobicity of the residues since it is the hydrophobic interaction in the proteins that trigger the accumulation of proteins [32]. The mutant PGA contains a single amino acid substitution where threonine residue is replaced with tyrosine. The hydrophobic feature of tyrosine present in mutant PGA and the hydrophilic feature of threonine maintained in wild-type PGA could explain the higher aggregation of protein in mutant PGA. Since the formation of inclusion bodies could result in improper folding of proteins to the correct structure, it reduces the activity of mutant PGA. Nevertheless, the specific activity value of mutant PGA expressed by arabinose induction is still higher than that expressed by IPTG induction, which indicates that the use of the same inducer generates the same pattern.
Many PGA mutants have been made in other research and the site-directed mutagenesis effects were investigated. While part of the mutants in these researches have increased PGA’s hydrolytic activity on penicillin-G, some of the mutants result in the decrease of PGA’s activity to hydrolyze penicillin-G. A study expressed three different PGA mutants recombinantly, and all of the mutants, especially βLeu24Phe, were able to increase the catalytic activity of the enzyme until 12-fold [48]. The research implemented the use of expressed chaperone and additional calcium chloride and thus encountered no trouble in generating functional PGA after the post-translational maturation process. In addition, the further purification steps in PGA recovery and the optimal binding of mutants to its substrate could be the other factors leading to the success. In contrast to the increased activity, the formation of three other PGA mutants in the Lys299 site has significant negative impacts on its activity due to the nonoptimal processing of PGA precursor [49]. Furthermore, although PGA mutant βAsn241Ala results in an activity-impaired enzyme even after optimal binding with the substrate, the inactive mutant provides more understanding of the role of the wild-type residue [26]. The low activity of mutant PGA due to the nonoptimal refolding could provide more insight into the function of the target residue in wild-type enzyme processing [50].
Further study on the ability and potential of βThr68Tyr mutant or the rest three potential mutants should be performed to confirm its hydrolytic activity, especially through more effective solubilization, refolding, and purification to recover more active PGA from inclusion bodies hence the enhanced enzymatic activity. More specific exploration of the protein processing and translocation is also important to generate the best expression condition that targets the complete folding of the protein thus producing a more soluble and functional protein. The further comparison between the mutant and wild-type PGA maturation could give more insight into the role of the native residue (βThr68) not only in enzymatic reaction but also in the post-translational folding process.
CONCLUSION
This study found 4 potential PGA mutants out of 36 mutants through homology modeling and molecular docking analysis. Other than having the stronger enzyme-substrate affinity, these mutants were predicted to be catalytically potential. The gene of one of these PGA mutants, βThr68Tyr, was synthesized and constructed in a pET-22b vector to be further explored. The transformation of the mutant recombinant plasmid into BL21(DE3) as the expression host generated mutant colonies which can grow in media culture and be induced for gene expression. The result shows that the mutant and wild-type PGA in soluble fraction obtained from the supernatant separated from sonicated cell suspension has better enzymatic activity than the solubilized and refolded PGA from insoluble fraction. Besides, arabinose has a greater potential as an inducer in comparison to IPTG based on the lower formation of inclusion bodies and higher value of enzymatic activity. Although the βThr68Tyr mutant has lower activity than wild-type PGA, the difference is not significant and the higher content of aggregated protein provides a new insight into the role of the original βThr68 residue in PGA folding and maturation. For a deeper understanding, optimization that includes the restoration of protein structure from aggregated form and recovery of purified PGA should be carried out in the future.
ACKNOWLEDGMENT
The authors would like to acknowledge the Final Project Recognition Grant Universitas Gadjah Mada Number 5075/UN1.P.II/Dit-Lit/PT.01.01/2023, Indonesia, for funding this research.
AUTHORS CONTRIBUTION
The research was designed by SSA, S, and P, and executed by SSA. Data were processed by SSA, S, and P. The manuscript was written and edited by SSA, S, and P.
CONFLICTS OF INTEREST
The authors declare there is no conflicts of interest with the current study.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Shewale JG, Sivaraman H. Penicillin acylase: enzyme production and its application in the manufacture of 6-APA. Process Biochem. 1989;24:146–54.
2. Srirangan K, Orr V, Akawi L, Westbrook A, Moo-Young M, Chou CP. Biotechnological advances on penicillin G acylase: pharmaceutical implications, unique expression mechanism and production strategies. Biotechnol Adv. 2013 Dec;31(8):1319–32. CrossRef.
3. Purwanto P, Sismindari S, Purwantini I. Production of penicillin G acylase (PGA) enzyme final progress report. Yogyakarta, Indonesia: Faculty of Pharmacy, University of Gadjah Mada; 2021 June. Contract No.: 87/E1/PRN/2020. Sponsored by the National Innovation Research Agency.
4. Tishkov VI, Savin SS, Yasnaya AS. Protein engineering of penicillin acylase. Acta Nat. 2010;2(3):47–61.
5. Shapovalova IV, Alkema WB, Jamskova OV, de Vries E, Guranda DT, Janssen DB, et al. Mutation of residue βF71 of Escherichia coli penicillin acylase results in enhanced enantioselectivity and improved catalytic properties. Acta Nat. 2009 Oct;1(3):94–8.
6. Yan Z, Huang B, Yang K, Anaman R, Amanze C, Jin J, et al. Enlarging the substrate binding pocket of penicillin G acylase from Achromobacter sp. for highly efficient biosynthesis of β-lactam antibiotics. Bioorg Chem. 2023 Jul;136:106533. CrossRef.
7. Oh SJ, Kim YC, Park YW, Min SY, Kim IS, Kang HS. Complete nucleotide sequence of the penicillin G acylase gene and the flanking regions, and its expression in Escherichia coli. Gene. 1987;56(1):87–97. CrossRef.
8. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. CrossRef
9. Alkema WB, Hensgens CM, Snijder HJ, Keizer E, Dijkstra BW, Janssen DB. Structural and kinetic studies on ligand binding in wild-type and active-site mutants of penicillin acylase. Protein Eng Des Sel. 2004 May;17(5):473–80. CrossRef.
10. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:296–303. CrossRef
11. Schrödinger L, DeLano W. PyMOL. 2020. Available from: http://www.pymol.org/pymol
12. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009 Dec;30(16):2785–91. CrossRef.
13. Alkema WB, Hensgens CM, Kroezinga EH, de Vries E, Floris R, van der Laan JM, et al. Characterization of the beta-lactam binding site of penicillin acylase of Escherichia coli by structural and site-directed mutagenesis studies. Protein Eng. 2000 Dec;13(12):857–63. CrossRef.
14. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–50. CrossRef.
15. Daggupati T, Naidu K, Pamanji R, Yeguvapalli S. Molecular screening and analysis of novel therapeutic inhibitors against c-Jun N-terminal kinase. Med Chem Res. 2017;26:1–7. CrossRef
16. Amin AM. Sequence analysis and gene expression of synthetic pac encoding penicillin G acylase (PGA) enzyme from Escherichia coli in host E. coli BL21(DE3) and E. coli HB101 [thesis]. Yogyakarta, Indonesia: University of Gadjah Mada; 2022.
17. Komari N, Hadi S, Suhartono E. [Protein modeling through homology modeling using SWISS-MODEL]. J Jejaring Mat Sains. 2020;2:65–70. CrossRef
18. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014 Jul;42:W252–8. CrossRef.
19. Wati W, Widodo GP, Herowati R. [Prediction of pharmacokinetics parameter and molecular docking study of antidiabetic compounds from Syzygium polyanthum and Syzygium cumini]. J Kimia Sains Aplikasi. 2020;23(6):189–95. CrossRef
20. Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi J, White SW, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009 Feb;49(2):444–60. CrossRef.
21. You L, Usher JJ, White BJ, Novotny J, inventor; Bristol Myers Squibb Co., assignee. Mutant penicillin G acylases. United States Patent US6403356B1. 2002 Jun 11.
22. Alkema WB, Dijkhuis AJ, De Vries E, Janssen DB. The role of hydrophobic active-site residues in substrate specificity and acyl transfer activity of penicillin acylase. Eur J Biochem. 2002 Apr;269(8):2093–100. CrossRef.
23. Alkema WB, Prins AK, de Vries E, Janssen DB. Role of alphaArg145 and betaArg263 in the active site of penicillin acylase of Escherichia coli. Biochem J. 2002 Jul 1;365(Pt 1):303–9. CrossRef.
24. Li Y, Song K, Zhang J, Lu S. A computational method to predict effects of residue mutations on the catalytic efficiency of hydrolases. Catalysts. 2021 Feb 22;11(2):2–14. CrossRef
25. Duggleby HJ, Tolley SP, Hill CP, Dodson EJ, Dodson G, Moody PC. Penicillin acylase has a single-amino-acid catalytic centre. Nature. 1995 Jan 19;373(6511):264–8. CrossRef.
26. McVey CE, Walsh MA, Dodson GG, Wilson KS, Brannigan JA. Crystal structures of penicillin acylase enzyme-substrate complexes: structural insights into the catalytic mechanism. J Mol Biol. 2001 Oct 12;313(1):139–50. CrossRef.
27. Morris GM, Lim-Wilby M. Molecular docking. Methods Mol Biol. 2008;443:365–82. CrossRef.
28. Asif A, Mohsin H, Tanvir R, Rehman Y. Revisiting the mechanisms involved in calcium chloride induced bacterial transformation. Front Microbiol. 2017 Nov 7;8:2169. CrossRef.
29. Zhou J, Li X, Xia J, Wen Y, Zhou J, Yu Z, et al. The role of temperature and bivalent ions in preparing competent Escherichia coli. 3 Biotech. 2018 May;8(5):222. CrossRef.
30. Malherbe G, Humphreys DP, Davé E. A robust fractionation method for protein subcellular localization studies in Escherichia coli. Biotechniques. 2019 Apr;66(4):171–8. CrossRef.
31. Carrió MM, Villaverde A. Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 2001 Jan 26;489(1):29–33. CrossRef.
32. Bhatwa A, Wang W, Hassan YI, Abraham N, Li XZ, Zhou T. Challenges associated with the formation of recombinant protein inclusion bodies in Escherichia coli and strategies to address them for industrial applications. Front Bioeng Biotechnol. 2021 Feb 10;9:630551. CrossRef.
33. Zeng H, Yang A. Quantification of proteomic and metabolic burdens predicts growth retardation and overflow metabolism in recombinant Escherichia coli. Biotechnol Bioeng. 2019 Jun;116(6):1484–95. CrossRef.
34. Kwon SB, Yu JE, Kim J, Oh H, Park C, Lee J, et al. Quality screening of incorrectly folded soluble aggregates from functional recombinant proteins. Int J Mol Sci. 2019 Feb 19;20(4):907. CrossRef.
35. Pan X, Yu Q, Chu J, Jiang T, He B. Fitting replacement of signal peptide for highly efficient expression of three penicillin G acylases in E. coli. Appl Microbiol Biotechnol. 2018 Sep;102(17):7455–64. CrossRef.
36. Jäger VD, Lamm R, Küsters K, Ölçücü G, Oldiges M, Jaeger KE, et al. Catalytically-active inclusion bodies for biotechnology-general concepts, optimization, and application. Appl Microbiol Biotechnol. 2020 Sep;104(17):7313–29. CrossRef.
37. Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng. 2005 Apr;99(4):303–10. CrossRef.
38. Kudou M, Ejima D, Sato H, Yumioka R, Arakawa T, Tsumoto K. Refolding single-chain antibody (scFv) using lauroyl-L-glutamate as a solubilization detergent and arginine as a refolding additive. Protein Expr Purif. 2011 May;77(1):68–74. CrossRef.
39. Yamaguchi H, Miyazaki M. Refolding techniques for recovering biologically active recombinant proteins from inclusion bodies. Biomolecules. 2014 Feb 20;4(1):235–51. CrossRef.
40. Mirhosseini SA, Latifi AM, Mahmoodzadeh HH, Seidmoradi R, Aghamollaei H, Farnoosh G. The efficient solubilization and refolding of recombinant organophosphorus hydrolyses inclusion bodies produced in Escherichia coli. J Appl Biotechnol Rep. 2019;6(1):20–5. CrossRef.
41. Torres LL, Ferreras ER, Cantero A, Hidalgo A, Berenguer J. Functional expression of a penicillin acylase from the extreme thermophile Thermus thermophilus HB27 in Escherichia coli. Microb Cell Fact. 2012 Aug 9;11:105. CrossRef.
42. Kasche V, Lummer K, Nurk A, Piotraschke E, Rieks A, Stoeva S, et al. Intramolecular autoproteolysis initiates the maturation of penicillin amidase from Escherichia coli. Biochim Biophys Acta. 1999 Aug 17;1433(1–2):76–86. CrossRef.
43. Ignatova Z, Mahsunah A, Georgieva M, Kasche V. Improvement of posttranslational bottlenecks in the production of penicillin amidase in recombinant Escherichia coli strains. Appl Environ Microbiol. 2003 Feb;69(2):1237–45. CrossRef.
44. López-Cano A, Sicilia P, Gaja C, Arís A, Garcia-Fruitós E. Quality comparison of recombinant soluble proteins and proteins solubilized from bacterial inclusion bodies. N Biotechnol. 2022 Dec 25;72:58–63. CrossRef.
45. Burgess RR. Refolding solubilized inclusion body proteins. Methods Enzymol. 2009;463:259–82. CrossRef.
46. Narayanan N, Hsieh MY, Xu Y, Chou CP. Arabinose-induction of lac-derived promoter systems for penicillin acylase production in Escherichia coli. Biotechnol Prog. 2006 May–Jun;22(3):617–25. CrossRef.
47. Lee SM. An efficient method for the release of recombinant penicillin G amidase from the Escherichia coli periplasm. J Life Sci. 2017, 27(10):1145–51.
48. Torres LL, Cantero A, del Valle M, Marina A, López-Gallego F, Guisán JM, et al. Engineering the substrate specificity of a thermophilic penicillin acylase from Thermus thermophilus. Appl Environ Microbiol. 2013 Mar;79(5):1555–62. CrossRef.
49. Lee H, Park OK, Kang HS. Identification of a new active site for autocatalytic processing of penicillin acylase precursor in Escherichia coli ATCC11105. Biochem Biophys Res Commun. 2000 May 27;272(1):199–204. CrossRef.
50. Aniqah SS, Molecular docking and site-directed mutagenesis of penicillin G acylase expressed in Escherichia coli BL21(DE3) and the effect on enzymatic activity [Thesis]. Yogyakarta, Indonesia: University of Gadjah Mada; 2022.
SUPPLEMENTARY FIGURE S1.
The sequence of mutant PGA (pgaEcβThr68Tyr) gene. The red arrow is pointing at the amino acid substitute, tyrosine, coded by the nucleotides at 1069–1071.