
INTRODUCTION
Lapatinib ditosylate monohydrate (LDM) is a potent inhibitor of ErbB-1 epidermal growth factor receptor and human epidermal growth factor receptor (ErbB-2) tyrosine kinases. It has been observed that LDM effectively inhibits the proliferation of cancer cells that exhibit overexpression of these two growth-promoting factors. TYKERB® 250 mg tablet, manufactured by Novartis Pharmaceuticals Corporation, is a commercially available LDM that has been approved by the US FDA for the treatment of advanced or metastatic breast cancer in 2007 [1]. The hepatic biotransformation of LDM is predominantly facilitated by CYP3A4 and CYP3A5 enzymes, while CYP2C19 and CYP2C8 isoenzymes play a minor role in the biotransformation process. Furthermore, it was established that LDM also serves as a substrate for both P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) efflux transporter proteins. Additionally, it also inhibits the functioning of P-gp, BCRP, and organic anion-transporting polypeptide 1B1 [2]. Although LDM exhibits high selectivity for tumors, its clinical effectiveness and safety have been challenging because of its inadequate water solubility and permeability. The characteristics highlighted above, i.e., limited bioavailability, high daily dose strength, and negative side effects, emphasize the importance of maintaining optimum therapeutic levels of LDM. This can be achieved by providing sufficient bioavailability to attain favourable patient outcomes [3,4].
The aqueous solubility of LDM is pH-dependent and can be affected by gastric acid secretion and intragastric pH changes. The solubility of the compound exhibits a significant decrease upon exposure to pH levels greater than 4.4. The oral absorption of a drug can be significantly affected by its solubility in water or physiological pH. This can result in restricted bioavailability or variability in the absorption process. The concern lies in the potential drug interaction that may lead to decreased LDM bioavailability when an acid-reducing agent is co-administered [5]. The clinical investigation conducted by Koch et al. [6] has reported a reduction in the aqueous solubility of LDM by a factor of 1,000 as the pH increases from 4 to 7 [6]. This decrease is in line with the reduction of the ionized fraction of LDM, which is a weak base having an acid dissolution constant (pKa) of 4.6 and 6.7. The pH of the gastrointestinal (GI) tract typically ranges from 1.6 in the stomach to 6.6 in the proximal small intestine, owing to the buffering properties of pancreatic and biliary secretions [7]. The properties imply that an elevation in pH results in reduction of aqueous solubility and subsequent decrease in absorption. The study conducted by Koch et al. [6] successfully attained test conditions by administering LDM at doses that are therapeutically relevant to patients who were concurrently using a proton pump inhibitor, a class of drugs which is commonly prescribed to cancer patients. In accordance with the findings of Koch et al. [6], it has been suggested that dosing LDM in a fasted state is advisable in order to attain a consistent therapeutic exposure, given the significant rise in LDM bioavailability and absolute variability. The consumption of a food resulted in a significant rise in the LDM area under the concentration-time curve (AUC) by 167% (2.67-fold) and maximum concentration (Cmax) by 142% (2.42-fold) [8].
Therefore, considering the above scenario the bioavailability of LDM is complex in both aspects. The ingestion of food is known to have a rapid buffering effect on gastric acid [9]. As a result, it is anticipated that the bioavailability of LDM would decrease in higher pH gastric environment. The Koch et al. [6] study’s findings indicate that LDM’s bioavailability is not significantly affected by pH and an increase in bioavailability was observed when administered with food. Weakly basic drugs tend to dissolve more readily in the fasted stomach as compared to weakly acidic drugs and forecasting the in vivo performance of poorly soluble weakly basic drugs is notably challenging [10]. Weakly basic compounds exhibit high solubility at low pH values due to their physicochemical properties. Consequently, significant inter- and intra-individual variability can be observed for these drugs because gastric pH is highly variable, even in the fasted state. The aqueous solubility of BSC class II and IV compounds can be misleading during drug development because the solubilizing capacity of the human intestinal environment is much higher than that of pure water or the buffers and dissolution media suggested by pharmacopeias’ and as a result, solubility problems may be overestimated [9,11].
Biorelevant media, such as fasted and fed state simulated intestinal fluid (FaSSIF and FeSSIF), have been designed to closely mimic human intestinal contents. These media, along with their variations, have significantly contributed to the advancement of biopharmaceutical in vitro tools, such as pH-shift or biphasic dissolution models. Physicochemical parameters such as surface tension, osmolality, viscosity, pH, and content of bile salts, phospholipids, and other amphiphiles can be used to characterize GI fluids. Amphiphiles present in intestinal fluids are crucial for solubilizing compounds that have poor solubility [12]. Using biorelevant dissolution media, numerous researchers have predicted the effect of dynamic variations in GI fluid characteristics in the fed, fasted, and bicarbonate buffer states on the intestinal dissolution of LDM [13,14]. Furthermore, while the solubility of LDM under specific pH conditions has been documented [15], the impact of pH shifts occurring in real-world physiological scenarios remains to be determined. In order to establish an in vitro–in vivo correlation, the results will be obtained using a micro-dissolution model comprising bio-relevant media, a physiologically acceptable clinical dose, and pertinent conditions, all of which should be taken into account. In this regard, the current investigation aims to analyse and enhance understanding regarding the molecular-level pH sensitivity of LDM, while also establishing a connection between the physicochemical characteristics of LDM in the presence of buffer and bio-relevant media. The principal aim of this investigation was to examine the influence of pH alteration on the release of LDM when exposed to conventional dissolution buffers and bio-relevant media, utilizing a micro-dissolution model.
MATERIALS AND METHODS
Chemicals and reagents
MSN Laboratories Pvt. Ltd., Hyderabad, provided a gift sample of LDM. Biorelevant (Croydon, Surrey, UK) supplied the media for fasted state-simulated gastric fluid (FaSSGF) and fasted state-simulated intestinal fluid (FaSSIF). Merck supplied buffers and salts i.e., potassium dihydrogen phosphate, sodium phosphate dibasic, hydrochloric acid, sodium chloride, and orthophosphoric acid. Milli-Q UV plus systems (Millipore Co., Bedford, MA) were used for purifying water. This study used analytical-grade compounds, solvents, and reagents for experiment.
Instrument and chromatographic conditions
The Shimadzu Prominence high-performance liquid chromatography (HPLC) system with LC-20 AT binary pumps, SPD-20A UV/VI’S detector, and SIL-20AC HT autosampler was used to implement the optimised chromatographic method for quantification of LDM. A Phenomenex C18 EVO (250 × 4.6 mm, 5 μ) column was used for chromatographic separation of LDM as a stationary phase with the mobile phase comprising of 10 mM ammonium acetate buffer of pH 4.5, acetonitrile, and methanol in the ratio 80:20 (The final mobile phase ratio was 60:40, buffer: organic phase) with a flow rate of 1 ml/minute at 253 nm wavelength using isocratic elution.
Software
Design of the experimental runs, analytical method optimization, and statistical analysis was carried out using Design Expert® software version 12.1 (Stat-Ease, Minneapolis, MN, Trail version).
Analytical design of experiment (DoE) method optimization
Defining analytical target profile (ATP) and critical analytical attributes (CAAs)
Establishing an ATP is the first stage in establishing a reversed-phase HPLC (RP-HPLC) method using the quality by design (QbD) methodology. The ATP includes (i) the separation of LDM and (ii) the quantification of LDM. A variety of critical method attributes may be selected in order to meet the ATP requirements. A critical quality attributes (CQAs) include retention time (Rt), tailing factor (Tf), height equivalent to a theoretical plate (HETP), spectral peak purity, resolution between impurities, resolution between the primary analyte and adjacent impurity, peak area, and assay, among others [16,17].
Risk assessment for screening of factors affecting the method
A risk assessment was performed as an essential element of the QbD methodology for method development, in order to determine the probability of potential risks or failures. In order to attain the desired outcome, a methodology involving the implementation of an Ishikawa fishbone diagram was employed to establish a causal connection between CAA and critical process parameters (CPPs) with the CQAs.
Method development using full factorial design
A factorial design comprising four factors was utilized for the initial screening of CAA like column, pH, and buffer. Four CAA/CPP were chosen for the screening which includes: A) pH of the buffer, B) percentage volume/volume of the aqueous mobile phase, C) column, and D) flow rate. The screening process was executed through the implementation of a three-level four-factorial design, which is presented in Table 1. A total of nine experiments were conducted with a uniform concentration of 10 μg/ml. The stated CQAs of Rt, theoretical plates (Tf), and HETP were used to analyse against the experimental data. A design model that best fits the data was generated and selected based on its significance using an analysis of variance (ANOVA) F test.
The final optimization was performed using Box-Behnken Design with A) pH of the buffer, B) the percentage volume per volume (% v/v) of the organic phase, C) the % v/v of modifier in a solvent, and D) the flow rate as CAA (Table 2). The experimental design consisted of 29 trial runs that employed a combination of CAA, as indicated in the design matrix. The experiments were conducted using drug concentration of 10 μg/ml of LDM and injection volume of 20 μl. The statistical techniques of ANOVA and response surface analysis were employed to further assess the model generated using DoE software.
Method validation
The developed analytical technique was validated for specificity, linearity, the limit of detection (LOD), the limit of quantification (LOQ), accuracy, and precision in accordance with the International Conference on Harmonization (ICH) Q2 (R1) [18].
Micro-dissolution pH shift experimentation
The micro-dissolution pH shift analysis of LDM was performed as per the our previously reported method [16,19,20]. Through a microdilution pH shift experiment using United States Pharmacopeia (USP) dissolution media and simulated bio-relevant media, the physiochemical changes and solubility of LDM in various pH transit was assessed. The pH levels for the two compartments of the in vitro micro dissolution test—gastric (FaSSGF buffer) and intestinal (FaSSIF buffer)—were 1.2, 6.5, and 6.8, respectively [21–23]. Prior to the experiment, all fluids were preheated to 37°C and degassed. The clinically equivalent dose of the LDM, 250 mg per 250 ml of water, was calculated to determine the target dose for the micro-dissolution setup. The dissolution experiment was then monitored for 20 minutes after the calculated quantity of LDM was added to 14 ml of pH 1.2 gastric medium (HCl/FaSSGF buffer). The pH was then shifted by adding 28 ml of pH 6.5 buffer (Phosphate/FaSSIF buffer) to the same setup upto and samples were withdrawn upto 180 minutes. In an alternate configuration, an analogous assembly was organized using 14 ml of Phosphate/FaSSIF buffer with a pH of 6.5. The dissolution process was executed for a duration of 20 minutes at pH 6.5. Subsequently, 28 ml of Phosphate/FaSSIF buffer with a pH of 6.8 was introduced in the same set-up to mimic the pH shift. The maintenance of sink conditions was achieved through the replacement of an equivalent volume of fresh buffer. The experimental fluids were mixed using magnetic stirrers rotating at a speed of 350 revolutions per minute, as directed by the assembly. The centrifugation of the samples was performed at a speed of 10,000 revolutions per minute. The resulting supernatants were analysed using the optimized RP-HPLC method, which was developed using the QbD strategy. The formula for calculating the “pH effect” is shown below as Equation 1.
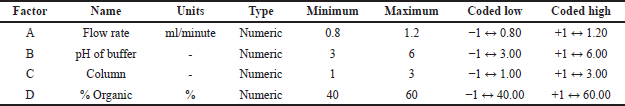 | Table 1. Selection of independent variables and their levels for factorial design. [Click here to view] |
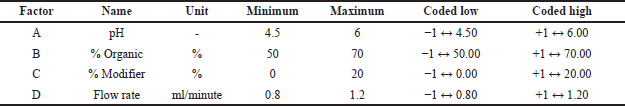 | Table 2. Selection of independent variables and their levels for optimization method using Box Behnken design. [Click here to view] |
RESULT AND DISCUSSION
Analytical DoE method development using factorial design
Experiments with distinct mobile phase solvents, buffers, and stationary phases were carried out to build a sensitive and effective analytical approach. Phosphate, acetate, and formate buffers were tested at pH 3–6. The effects of four independent technique factors, mobile phase pH (X1), % organic in the mobile phase (X2), stationary phase (X3), and flow rate (X4), on three LDM response variables, drug Rt (R1), Tf (R2), and HETP (R3), were examined using a four-factor three-level factorial design. The proposed chromatographic experimental conditions via DoEs were carried out using HPLC, and the findings were evaluated by statistical analysis using Design Expert 12.0 software.
The initial risk assessment by the analysis of the normal plot and perturbation plot revealed favourable interactions between CPP such as pH, % organic, flow rate, and CQA of Rt, Tf, and HETP. Additionally, the factorial screening design underwent statistical evaluation using ANOVA. The statistical model demonstrated significance when analyzed using a two-factor polynomial model fit design. The variable X1, which represents the pH of the mobile phase, exhibited a statistically significant influence on the responses Rt (R1) and HETP (R3). The variables of flow rate, column, and % organic was observed to have a significant impact on the responses R2 and R3. The outcomes of the factorial screening design utilizing ANOVA are presented in Table 3. The statistical parameters, namely p-value, F-value, R2, and adjusted R2, exhibited a satisfactory level of concurrence and demonstrated a noteworthy interplay between CPP and CQA of the analytical approach (Fig. 1). The utilization of 3D interaction plots facilitated the examination of the interplay between the dependent and independent variables. The response R1 (Rt) of LDM was significantly influenced by independent parameters, namely the pH of the mobile phase and the columns utilized. Likewise, the response R2 (Tf) of LDM exhibited a significant dependence on both the flow rate and the % of organic solvent, as observed in response R2. The study indicates that the response factors Rt and HETP do not exhibit any statistically significant impact in relation to independent variables, namely pH and flow rate (Fig. 2). The screening design results indicated that the dependent variables, namely Tf and HETP, remained constant within the pH range of 4.5–6 and the organic percentage range of 50%–60%. The selection of the C18 column was based on the impact of the independent variable, column, on the response variable R3.
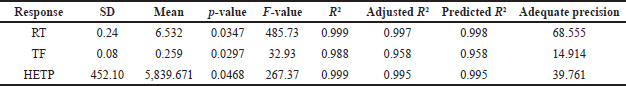 | Table 3. ANOVA results for factorial screening design. [Click here to view] |
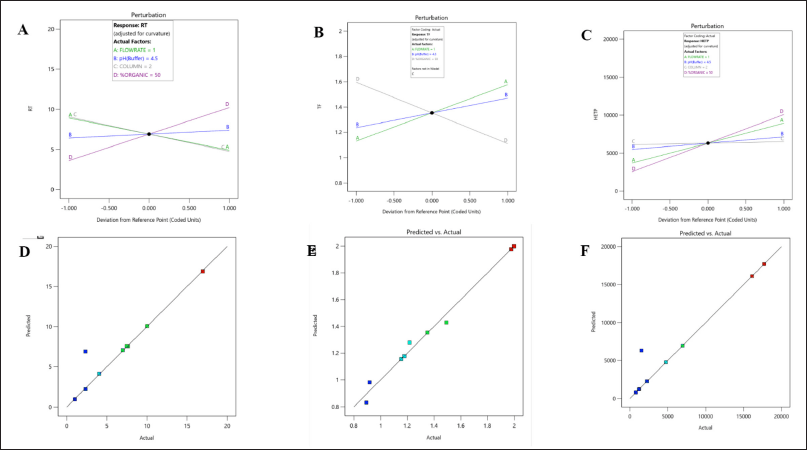 | Figure 1. Perturbations and predicted versus actual plot interactions for dependant factors A, D; (R1) Rt, B, E; (R2) Tf and C, F; (R3) HETP of LDM by three level four factorial design. [Click here to view] |
Analytical DoE method optimization using Box-Behnken design
The utilization of the randomized quadratic Box-Behnken design was extended to facilitate the optimization of pH and the % of organic ratio with the aid of a modifier. The independent variables were subjected to a risk assessment using a normal probability perturbation plot and an actual versus predicted plot. The perturbation plot was found to fall within the acceptance criteria. The ANOVA-based statistical analysis reveals that the % of modifiers and organic content have a significant impact (p < 0.001) on the responses R1 and R2. Conversely, the pH of the mobile phase and flow rate exhibited no significant impact on either of the responses. The ANOVA results for the quadratic model, which was determined to be the best fit, are presented in Table 4. The ANOVA predicted that the response R1 would exhibit a similar interaction profile for independent variables such as % organic, % modifier, and flow rate, as evidenced by the 3D counterplots of the analysis. The 3D interaction plot presented illustrates the relationship between the response R1 (Rt) and the % of organic and modifier, as well as the pH of the mobile phase. Nevertheless, Tf has demonstrated the most noteworthy interaction with the percent modifier and percent organic (Fig. 3). The method operable design region (MODR) value obtained under optimized conditions indicated that the design space for pH ranged from 4.5 to 6, while the percentage of organic and modifier fell within the ranges of 55%–60% and 10%–20%, respectively. The study determined that the optimized method had a desirability score of 1. Working within the operatable region of the design space is likely to result in a satisfactory yield (Fig. 4). Table 5 presents a summary of the observed and forecasted outcomes for the reactions obtained through the optimized chromatographic technique for LDM.
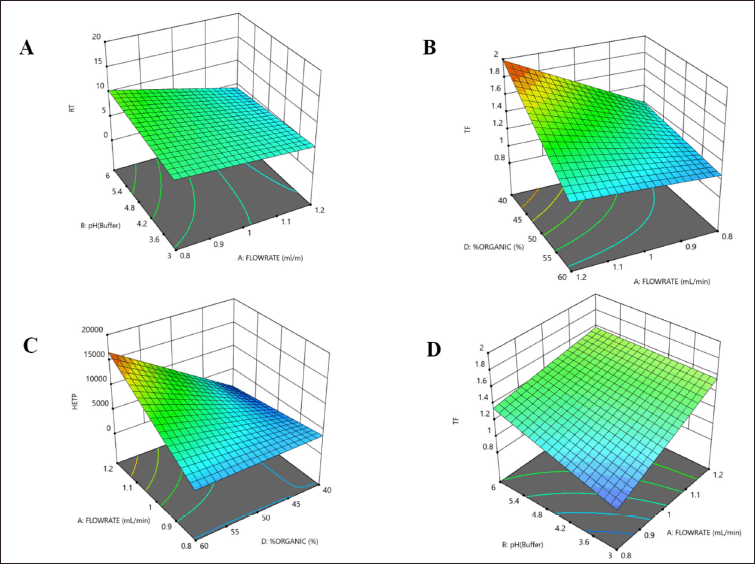 | Figure 2. 3D counter plot of interactions for dependant factors (A); (R1) Rt, (B, C); (R2) Tf and (D); (R3) HETP of LDM by three level four factorial design. [Click here to view] |
Method validation
The indicated parameters’ findings were assessed, and the method was validated in accordance with the ICH Q2(R1) guidelines. As the % relative standard deviation (RSD) of the standards was less than 2%, the system appropriateness was well within the permitted range. The concentration versus peak area calibration curve was plotted as shown in Figure 5 after the linearity for LAP was determined. Through statistical analysis, the correlation coefficient and intercept were obtained with a regression equation of y = 29,336X–1,450.9. Figure 5 illustrates the correlation coefficient (r2), which was found to be 0.9999. According to Table 7, the LOQ was 0.494 μg/ml and the LOD was 0.163 μg/ml. As indicated in Table 6, the mean percent recovery was found to be accurate and within the range of 98%–102%. The offered method’s repeatability and intermediate precision were used to determine its accuracy. According to Table 7, the %RSD was calculated to be less than 2%.
 | Table 4. ANOVA results for optimised method using Box-Behnken design. [Click here to view] |
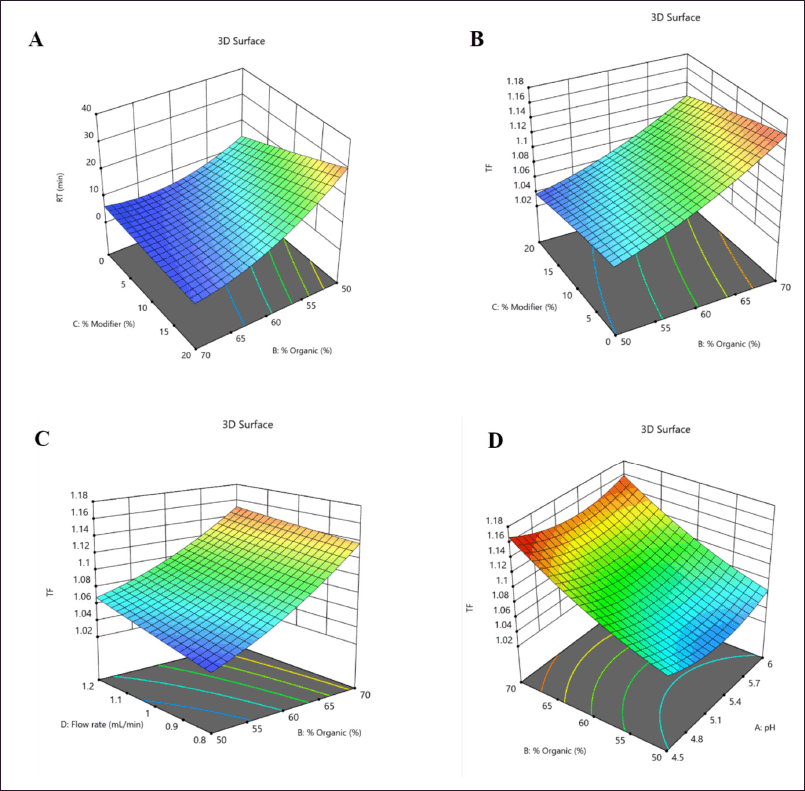 | Figure 3. 3D counter plot of interactions for dependant factors (A); (R1) Rt and (B, C, D); (R2) Tf of LDM by Box-Behnken design. [Click here to view] |
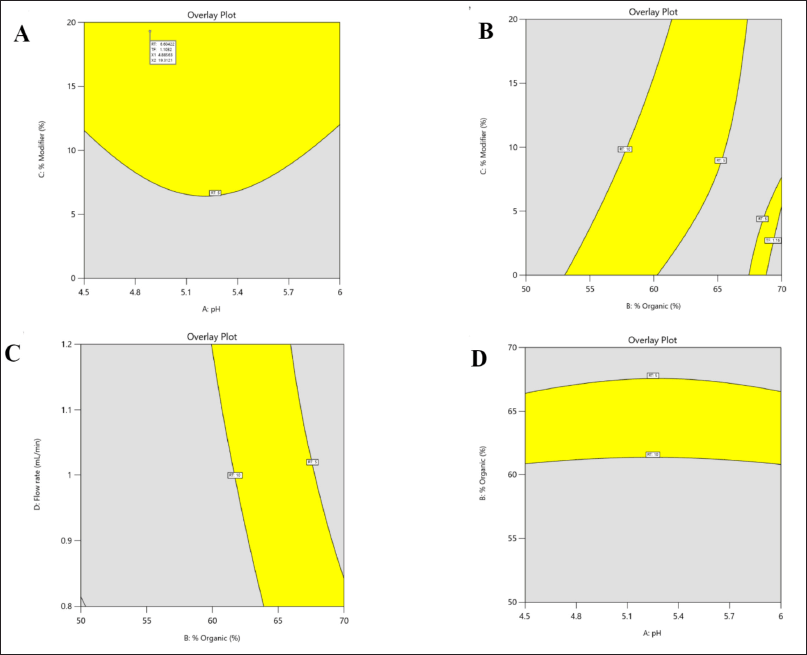 | Figure 4. Design space of method operatable region for LDM with independent factors (X1) pH, (X2) % organic, (X3) % organic modifier and (X4) flow rate. [Click here to view] |
 | Table 5. Predicted and actual point prediction at two-sided 95% confidence interval. [Click here to view] |
Micro-dissolution pH shift experimentation
The intrinsic dissolution rate and pH-dependent solubility of LDM, a drug with low solubility, were evaluated through the use of USP dissolution buffers and bio-relevant media (FaSSGF-FaSSIF) in a micro-dissolution model. This was done to investigate the pH shift transition from the gastric to intestinal phase. The impact of pH on LDM solubility and its dissolution profile in USP buffer is demonstrated in Figure 6, which displays data for HCl pH 1.2 and Phosphate buffer pH 6.5. The initial drug concentration was dissolved in a buffer solution with a pH of 1.2, in which 0.0164 mg/ml of LDM was found to be soluble. Upon modifying the pH of the experimental system from 1.2 to 6.5, an instantaneous drop in the solubility of the drug was observed, resulting in a value of 0.0040 mg/ml. According to the report, LDM exhibited solubility values of 0.008 and 0.006 mg/ml in the intestinal phase set of phosphate buffer at pH 6.5 and 6.8, respectively. The observed reduction in drug solubility subsequent to the pH alteration underscores the critical role of pH in drug solubility and illustrates the pH-dependent nature of the drug. Likewise, in a micro-dissolution setup utilizing bio-relevant media, LDM was observed to exhibit minimal solubility in an acidic milieu, specifically FaSSGF at a pH of 1.2. Following a duration of 20 minutes, a significant reduction in the solubility of LDM during GI transit was observed, plausibly due to the precipitation of the more soluble acidic di-tosylate salt. The impact of a pH shift on the solubility and dissolution profile of LDM in bio-relevant media is depicted in Figure 7, where the transition is made from FaSSGF pH 1.2 to FaSSIF pH 6.5. The solubility of the compound was initially measured to be 0.0127 mg/ml; however, it underwent a significant increase to 0.0291 mg/ml following the pH alterations. During the transition from intestinal phase shift of FaSSIF pH 6.5 to FaSSIF pH 6.8, the solubility of LDM was found to be 0.0337 mg/ml and remained constant until it reached saturation equilibrium. Upon changing the pH to 6.8, the solubility of LDM decreased to 0.0289 mg/ml and this value was maintained for a duration of 180 minutes.
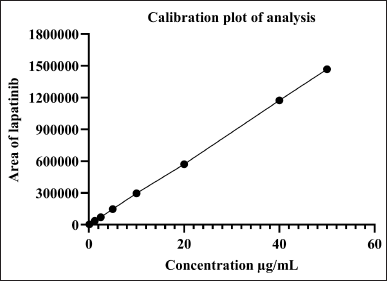 | Figure 5. Calibration plot of analysis for LDM. [Click here to view] |
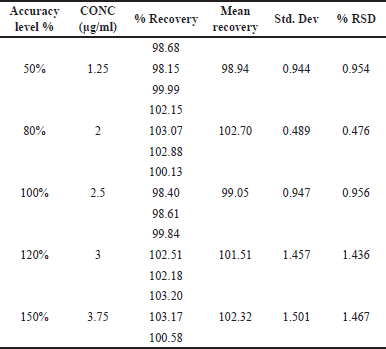 | Table 6. Accuracy of LDM at 5 levels. [Click here to view] |
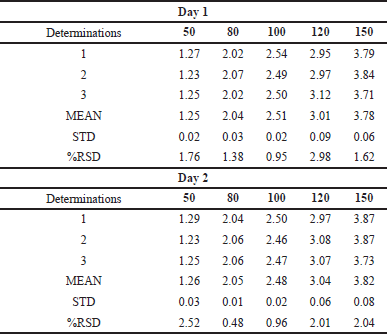 | Table 7. Intraday-interday precision of LDM at 5 levels. [Click here to view] |
In gastric bio-relevant media, the drug exhibits limited solubility with a release of up to 20% during the first 20 minutes of dissolution. The dissolution of LDM was observed to be significantly faster in the intestinal fluids of FaSSIF pH 6.5, with a release of 75%–80% within 180 minutes. LDM exhibits poor water solubility and high lipophilicity, with a logP value of 5.66. It also displays weak basicity, with a pKa of approximately 3.7, and weak acidity, with a logP value of 5.66. Under highly acidic aqueous conditions, LDM has the ability to undergo ionization and solubilization. Various researches have been conducted to investigate the solubility of the unpolluted medication. These studies have revealed that the amount of drug dissolution at pH 2 is notably low [7,9,13]. Koch et al.’s [6] study on the pH-dependent interaction of LDM in humans revealed that despite the observed decrease in aqueous solubility in vitro, the reduction in LDM absorption was not significant. The observed discrepancy in drug efficacy could potentially be attributed to the interplay between the physicochemical properties of the drug and the environmental conditions within the intestinal lumen. The hydrophobic nature of LDM promotes the segregation of amphoteric bile salts into micelles within the enterocytes’ wall or the duodenum. This mechanism serves to counterbalance the reduction in aqueous solubility that occurs at elevated pH levels. The inclusion of amphiphiles within the GI tract significantly amplifies the solubility of numerous compounds that exhibit poor water solubility. However, it is crucial to take into account the impact of distinct amphiphiles on solubility, as well as the partitioning of the drug into amphiphilic micelles, as these factors can influence the absorption rate and degree [8,15,24].
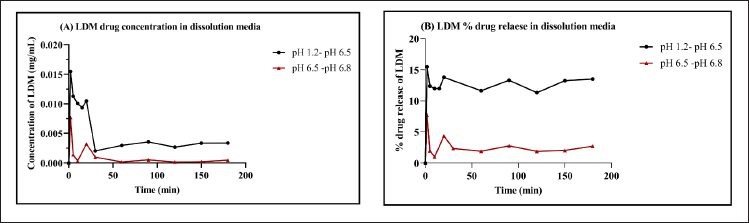 | Figure 6. Effect of pH shift on solubility of LDM for (A) HCl pH 1.2→Phosphate pH 6.5 and (B) % drug release by in vitro micro dissolution. [Click here to view] |
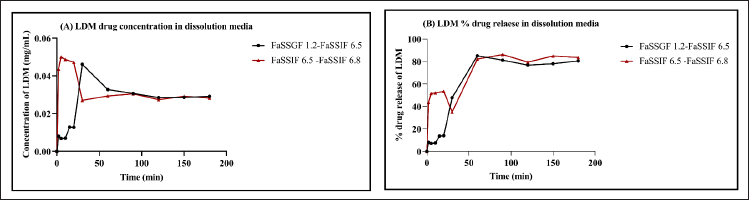 | Figure 7. Effect of pH shift on solubility of LDM for (A) FaSSGF 1.2→FaSSIF pH 6.5 and (B) % drug release by in vitro micro dissolution. [Click here to view] |
Additionally, the ratio of the dissolution AUC profiles (AUC of pH 6.5 to pH 6.8/AUC of pH 1.2 to pH 6.5) was used to evaluate the risk of in vitro pH impact. In a human clinical study with and without an acid lowering agent, LDM’s predicted clinical pH impact ratio was reported as 0.784 and 0.743 for Cmax and AUCinf, respectively. The in vitro pH-effect risk obtained from the micro-dissolution experiment has values of 3.787 and 0.8010, respectively, which indicate the use of USP buffer and bio-relevant media. The clinical effects of pH and in vitro pH are all directly related to one another. In reality, the pH influence is more substantial the greater the ratio. When the pH ratio is 1, there are no clinical consequences of pH. There haven’t been many solubility investigations where the solubility in human subjects has been directly compared to the solubility in biorelevant media mimicking human digestive fluids. Through enhanced wetting and solubilization by additives like surfactants and/or lipids, intestinal simulated bio-relevant fluids frequently have a significant impact on the apparent solubility of compounds with solvation-limited solubility, i.e., molecules with poor interaction with water [25].
The solubility and consequent absorption of a drug from the GI tract are largely influenced by the composition and volume of the GI fluids, as well as the micellar concentration. The administration of cancer therapies through oral means has prompted the need to closely examine the factors that contribute to fluctuations in drug exposure, which ultimately impact the delicate balance between therapeutic effectiveness and harmful side effects. Conducting an extensive physiochemical analysis utilizing bio-relevant media is deemed as the most dependable approach to attain uniform systemic exposure while administering targeted drug therapy through oral route for a prolonged period.
CONCLUSION
The current study outlined the use of biorelevant FaSSGF and FaSSIF in an in vitro pH shift micro-dissolution model to predict the solubility change of LDM in the GI system. The use of FaSSIF-based buffers led to more precise predictions of the supersaturation and dissolution rate of the pure weakly basic model compound LDM when compared to normal buffers. Our prediction for the pH shift effect has successfully integrated the dynamic changes in GI fluid properties and their implications on the in vivo dissolution of LDM. A better understanding of the impact of food and acid-reducing substances on the exposure of poorly water-soluble drugs may be made possible by the conduct of such an assessment.
ACKNOWLEDGMENT
The authors would like to acknowledge Manipal College of Pharmaceutical Sciences, Manipal and Manipal Academy of Higher Education Manipal for providing resources and research facilities for conducting the research work.
AUTHOR CONTRIBUTIONS
Conceptualization: Anithakumari Uttam Singh Rajpurohit, Prajakta Patil; Data curation: Anithakumari Uttam Singh Rajpurohit, Prajakta Patil, Mrunal Desai; Formal analysis: Prajakta Patil, Mrunal Desai;Funding acquisition: Jagadish Puralae Channabasavaiah; Investigation: Jagadish Puralae Channabasavaiah; Methodology: Anithakumari Uttam Singh Rajpurohit, Prajakta Patil, Mrunal Desai; Project administration: Jagadish Puralae Channabasavaiah; Resources: Jagadish Puralae Channabasavaiah; Supervision: Jagadish Puralae Channabasavaiah; Visualization: Jagadish Puralae Channabasavaiah; Validation: Prajakta Patil, Jagadish Puralae Channabasavaiah; Writing–original draft: Prajakta Patil; Writing–review and editing: Mrunal Desai, Jagadish Puralae Channabasavaiah.
FINANCIAL SUPPORT
The authors received no financial support for the research of this article.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
ETHICAL APPROVAL
The study does not have involvement of experiments on humans or animals.
DATA AVAILABILITY
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Oakman C, Pestrin M, Zafarana E, Cantisani E, di Leo A. Role of lapatinib in the first-line treatment of patients with metastatic breast cancer. Cancer Manag Res. 2010;2(1):13–25. CrossRef
2. Karbownik A, Sobanska K, Plotek W, Grabowski T, Klupczynska A, Plewa S, et al. The influence of the coadministration of the p-glycoprotein modulator elacridar on the pharmacokinetics of lapatinib and its distribution in the brain and cerebrospinal fluid. Invest New Drugs. 2020;38(3):574–83. CrossRef
3. Hu XY, Lou H, Hageman MJ. Preparation of lapatinib ditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int J Pharm. 2018;552(1–2):154–63. CrossRef
4. Jede C, Wagner C, Kubas H, Weber C, Weigandt M, Koziolek M, et al. Application of an automated small-scale in vitro transfer model to predict in vivo precipitation inhibition. Int J Pharm. 2019;565(April):458–71. CrossRef
5. U.S. Food and Drug Administration (FDA). Tykerb (lapatinib) [Bula]. 2007;1–25.
6. Koch KM, Im YH, Kim SB, Urruticoechea Ribate A, Stephenson J, Botbyl J, et al. Effects of esomeprazole on the pharmacokinetics of lapatinib in breast cancer patients. Clin Pharmacol Drug Dev. 2013;2(4):336–41. CrossRef
7. Fink C, Sun D, Wagner K, Schneider M, Bauer H, Dolgos H, et al. Evaluating the role of solubility in oral absorption of poorly water-soluble drugs using physiologically-based pharmacokinetic modeling. Clin Pharmacol Ther. 2020 107(3):650–61. CrossRef
8. Lewis LD, Koch KM, Reddy NJ, Cohen RB, Lewis NL, Whitehead B, et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J Clin Oncol. 2009;27(8):1191–6. CrossRef
9. Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12(3):397–406. CrossRef
10. Kleberg K, Jacobsen J, Müllertz A. Characterising the behaviour of poorly water soluble drugs in the intestine: application of biorelevant media for solubility, dissolution and transport studies. J Pharm Pharmacol. 2010;62(11):1656–68. CrossRef
11. Kádár S, Csicsák D, Tozsér P, Farkas A, Pálla T, Mirzahosseini A, et al. Understanding the pH dependence of supersaturation state—a case study of Telmisartan. Pharmaceutics. 2022;14(8):1–14. CrossRef
12. Jamil R, Polli JE. Prediction of in vitro drug dissolution into fed-state biorelevant media: contributions of solubility enhancement and relatively low colloid diffusivity. Eur J Pharm Sci. 2022;173(March):106179. CrossRef
13. Kiyota T, Ando Y, Kambayashi A. Dynamic changes in gastrointestinal fluid characteristics after food ingestion are important for quantitatively predicting the in vivo performance of oral solid dosage forms in humans in the fed state. Mol Pharm. 2023;20(1):357–69. CrossRef
14. Jede C, Wagner C, Kubas H, Weigandt M, Weber C, Lecomte M, et al. Improved prediction of in vivo supersaturation and precipitation of poorly soluble weakly basic drugs using a biorelevant bicarbonate buffer in a gastrointestinal transfer model. Mol Pharm. 2019;16(9):3938–47. CrossRef
15. Fink C, Lecomte M, Badolo L, Wagner K, Mäder K, Peters SA. Identification of solubility-limited absorption of oral anticancer drugs using PBPK modeling based on rat PK and its relevance to human. Eur J Pharm Sci. 2020;152(June):105431. CrossRef
16. Patil PH, Desai M, Rao RR, Mutalik S, Shenoy GG, Rao M, et al. Assessment of pH-shift drug interactions of palbociclib by in vitro micro-dissolution in bio relevant media: an analytical QbD-driven RP-HPLC method optimization. J Appl Pharm Sci. 2022;12(5):78–87.
17. Ramesh A, Channabasavaish JP, Jhawar V, Das P, Patil P, Mutalik S. Maraviroc oral disintegration tablet: analytical design of experiments (DoE) for assessment and comparison of in vitro dissolution profiles. Curr Pharm Anal. 2021;18(4):427–36. CrossRef
18. Harron DWG. Technical requirements for registration of pharmaceuticals for human use: the ICH process. Textb Pharm Med. 2013;1994(October 1994):447–60. CrossRef
19. Kolasani DD, Desai M, Patil P, Jagadish PC. Evaluation of pH dependent solubility and examination of variation in pharmacokinetic properties of alectinib: a quantitative study by implementing integrated quality by design approach for RP-HPLC method development and optimization. Indian J Pharm Educ Res. 2022;56(4):1219–25. CrossRef
20. Desai M, Patil PH, Rao RR, Shenoy GG, Rao M, Mutalik S, et al. Should the use of acid reducing agents in conjunction with ribociclib be avoided? An integrated QbD approach for assessment of pH-mediated interaction. J Chromatogr Sci. 2022;1–7. CrossRef
21. Varma MV, Gardner I, Steyn SJ, Nkansah P, Rotter CJ, Whitney-Pickett C, et al. PH-dependent solubility and permeability criteria for provisional biopharmaceutics classification (BCS and BDDCS) in early drug discovery. Mol Pharm. 2012;9(5):1199–212. CrossRef
22. Zhu AZX, Ho MCD, Gemski CK, Chuang BC, Liao M, Xia CQ. Utilizing in vitro dissolution-permeation chamber for the quantitative prediction of pH-dependent drug-drug interactions with acid-reducing agents: a comparison with physiologically based pharmacokinetic modeling. AAPS J. 2016;18(6):1512–23. CrossRef
23. Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, et al. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci. 2020;109(3):1380–94. CrossRef
24. Fagerberg JH, Tsinman O, Sun N, Tsinman K, Avdeef A, Bergström CAS. Dissolution rate and apparent solubility of poorly soluble drugs in biorelevant dissolution media. Mol Pharm. 2010;7(5):1419–30. CrossRef
25. Fotaki N, Vertzoni M. Biorelevant dissolution methods and their applications in in vitro in vivo correlations for oral formulations. Open Drug Deliv J. 2010;4(SPEC. ISSUE 1):2–13. CrossRef