
INTRODUCTION
Amide cinnamate and cinnamic acid-related compounds constitute a class of anti-cancer agents, which include cinnamic acid, cinnamate ester, prenylated cinnamic acid, cinnamoyl ester, cinnamoyl toxoids, carbamoyl imidazole cinnamate, cinnamoyl pyrrole, cinnamoyl pyrrolidine, cinnamoyl triazole, cinnamoyl quinazoline, cinnamoyl pyrimidine, cinnamoyl piperazine, cinnamic-hydroxamic acid, and cinnamoyl hydrazides [1–16]. Structural modification plays a crucial role in the discovery of derivatives with specific biological activities [17]. Many amide cinnamate compounds have been synthesized and evaluated for their anti-cancer activity. 2-methyl cinnamide, isolated from a Streptomyces griceoluteus, demonstrated significant anti-invasive or anti-metastatic effects. When used as pretreatment for malignant melanoma cells (C8161 and A375 M) in vitro, this compound exhibited a dose and time-dependent reduction in invasion (IC50 = 12.5 μg/ml) [1]. Amide cinnamate, cinnamic acid, ester cinnamate, and other related derivatives represent important drug candidates for anti-cancer therapy; however, their potential as anti-cancer agents is still underutilized. These compounds exert various anti-cancer effects, including cytotoxic, cytostatic, anti-tumors, anti-leukemic, anti-angiogenetic, anti-proliferative, inhibition of different enzymes, and DNA damage.
Matrix metalloproteinases (MMPs) or matrix metalloproteinases are a type of endopeptidase consisting of pro-peptides and catalytic domains, which rely on zinc for their function [18]. They are classified as collagenase, stromelysins, or gelatinases based on their preference for different substrates [19]. Several types of cancer, including prostate cancer, mammary gland cancer, and alveolar cancer, have been found to express MMP-9. Natural and synthetic inhibitors of MMP-9 protein are useful for cancer treatment. Recently, cinnamic acid derivatives have been identified as effective and specific inhibitors of MMP-9 for cancer treatment [20].
Previously, we conducted a study in which we designed, synthesized, and examined the molecular docking of several novel bioactive derivatives of methyl trans-cinnamate [21,22]. In this study, we used methyl trans-cinnamate as the lead compound. We removed the methyl ester group and replaced it with an amide group, after which we investigated the anti-cancer activity of these amide cinnamate derivatives using the Michigan Cancer Foundation-7 (MCF-7) breast cancer cell line in vitro. In addition, we performed in silico studies to evaluate the potential of these compounds as MMP-9 inhibitors. This study is particularly useful for exploring molecular interactions, predicting the behavior of complex systems, analyzing large datasets, and making predictions about the outcomes of experiments or observations that might be time-consuming or costly to perform in a laboratory or in vivo setting. In silico studies are commonly employed in fields such as drug discovery, protein structure prediction, molecular dynamics simulations, genomics, and systems biology. These studies can provide valuable insights and guide experimental research by narrowing down potential areas of interest and helping researchers make informed decisions about which experiments to pursue further. Finally, we assessed the correlation between the in vitro and in silico anticancer activity testing of the cinnamate amide derivatives.
MATERIALS AND METHODS
Materials
For this study, we utilized compound 1, which is methyl trans-cinnamate isolated from Alpinia malacensis, as well as compound 2, trans-cinnamic acid that was obtained through the hydrolysis of compound 1. We also synthesized compounds 3-9 through the amidation reaction between alkyl amines and cinnamic acid [23]. All of these compounds (compounds 1-9) were subjected to spectroscopic characterization using a variety of techniques, including UV spectrophotometry, Fourier-transform infrared spectroscopy, 1H-NMR, 13C-NMR, gas chromatography–mass spectrometry, and electrospray ionization quadrupole time-of-flight mass spectrometry. Alamar Blue (Invitrogen™, DAL1025, Oregon, US), Dulbecco’s modified Eagle’s red medium (Gibco, DMEM, Grand Island, US), Fetal Bovine Serum (Gibco, US).
In vitro anti-cancer activity
In order to assess the anti-cancer activity of compounds 1-9, an in vitro test was conducted using the breast cancer cell line MCF-7. This test was performed by measuring cell viability using AlamarBlue. The cells were cultured using Dulbecco’s modified Eagle’s red medium supplemented with 10% v/v fetal bovine serum and 1% antibiotic-antimycotic and were incubated at 37°C with 5% CO2. The cells were seeded in each well at a concentration of 5 × 104 cells/ml and incubated for 3 hours at 37°C under 5% CO2. The samples and tamoxifen were added to the wells at concentrations of 100 μl/ml and 40 μl/ml, respectively, and were incubated for 24 hours. After this, 10 μl of AlamarBlue reagent was added to each well and the plates were incubated for 3 hours. The fluorescence was then measured at 560 nm excitation and 590 nm emission using a Varioskan microplate reader [24–26].
Molecular docking study
AutoDock 4.2 program was used to conduct molecular docking studies on amide cinnamate derivatives as potential anti-cancer agents. The purpose of this study was to gain insight into the binding mode of interaction between these derivatives and cancer receptors, particularly the α-estrogen receptor obtained from protein data bank (PDB) with PDB ID: 4WZV, which underwent external validation. The amide cinnamate derivatives were converted into a 3D structure, minimized, and converted into mol2 format before being saved as pdbqt. The receptor was prepared by removing water, adding hydrogen atoms, and providing Gasteiger partial charge before being saved as pdbqt. A grid file was created based on the native ligand active site pocket with dimensions of 40 × 40 × 40 and coordinates of x = −0.769, y = −0.934, and z = 30.686. A grid file and a docking file were generated in gpf and dpf formats, respectively. The results of the docking study were determined by the binding energy value between the receptor and ligand based on the lowest energy [27–29].
RESULTS AND DISCUSSION
The development of new drugs is crucial in the search for effective treatments to inhibit the growth of cancer cells. Drug development research typically involves in vitro studies using cancer cells and in silico prediction of structure-activity mechanisms for newly synthesized compounds. Quantitative structure and activity relationship (QSAR) is a useful tool to examine the effect of functional groups on the biological activity of chemical compounds. Understanding QSAR can increase the potential biological activity of a chemical, leading to the design of compounds with higher efficacy in inhibiting the growth of cancer cells. Design strategies for new drug compounds should consider factors such as polarity, steric factors, log P values, Lipinski’s rule, and a variety of functional groups.
Table 1 lists the design of amide cinnamate derivatives (compounds 1-9) and their respective log P, molecular weight, and molecular formula. The evaluation of in vitro and in silico anti-cancer activity of amide cinnamate derivatives (compounds 1-9) was conducted to see the QSAR. Figure 1 shows the effect of amide cinnamate derivatives concentration on their cytotoxicity to MCF-7. The concentration of 100 µg/ml was used as an initial screening for in vitro anti-cancer cell line activity of extracts, fractions, or compounds in our lab. If activity was observed, testing at lower concentrations in this study was tested at 40 µg/ml. Almost all amide cinnamate derivatives at the concentration of 100 µg/ml had strong inhibition to the MCF-7 cell growth. This observation indicated that at this concentration, almost all the amide cinnamate derivatives possessed potential activity as anti-cancer. Nevertheless, when the sample concentration was lowered to 40 µg/ml, most of the compounds with hydroxyl substituents in the carboxylate group completely lost their cytotoxicity to MCF-7 (compound 2). Moreover, the same observation occurred in the amide cinnamate derivatives with short-chain alkyl amine substituents in the carboxylate group (compounds 4 and 6). When the substituent of the carboxylate group was long-chain alkyl amine (compound 7) or alkyl amine with benzene ring (compounds 8 and 9), percentage inhibition to MCF-7 was more than 40%. Molecular weight and log P value were also found to be important aspects. From the data shown in Table 1, compounds 9, 7, and 8 demonstrated good inhibition. The highest cytotoxicity was displayed by compound 8 (Fig. 1). The results obtained in this study indicated that cytotoxicity or potential anticancer activity mainly contributed to the amide structure present in the compound. This observation was in agreement with the results reported on the natural amide cinnamide isolated from Streptomyces griseoluteus [1]. The microscopic observation of control cells (no sample addition) and cytotoxicity (40 μg/ml sample concentration) of compound 1 (moderate activity) and compound 8 (highest activity) are shown in Figure 2. This result clearly showed that most cells treated with compound 8 were unattached to the well.
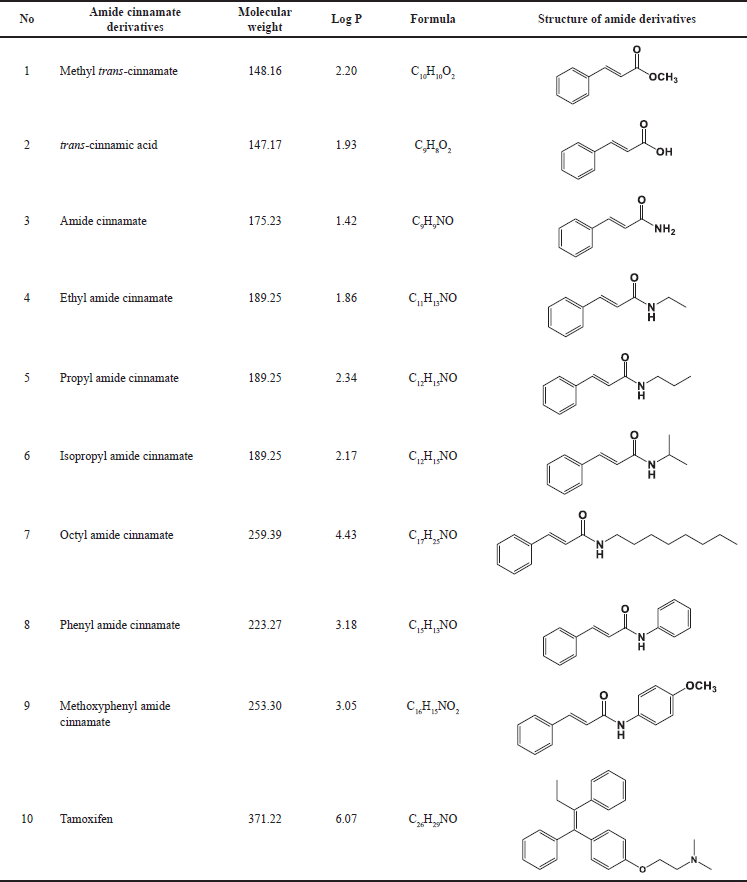 | Table 1. Tested samples of amide cinnamate derivatives. [Click here to view] |
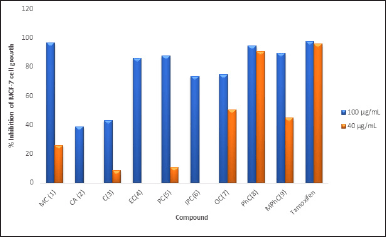 | Figure 1. Percentage inhibition of amide cinnamate derivatives at sample concentration of 100 and 40 μg/ml to MCF-7 cell growth. [Click here to view] |
 | Figure 2. Microscopic observation of cytotoxic activities of amide cinnamate derivatives against MCF-7 cancer cells line. (a) Control cells (untreated), treated cells with 40 μg/ml with (b) methyl trans-cinnamate (compound 1) and (c) phenyl amide cinnamate (compound 8). [Click here to view] |
Table 2 summarizes the molecular docking simulation results of compounds 1-8. The results of this docking study were expressed by the amount of energy required to bind the mode interaction in the 4 WZV receptor. The lower the energy required, the more potential the compound is in binding to the binding mode interaction with the receptor. The docking protocol was validated by re-docking the native ligand compound into the 4 WZV receptor pocket. Validation is considered to be quite feasible if the root mean square deviation value from the re-docking results is below 2 Å. Phenyl amide cinnamate (8), methoxyphenyl amide cinnamate (9), and octyl amide cinnamate (7) showed good binding mode binding at the 4 WZV receptor active site. Several amide cinnamate compounds were designed to identify a new set of molecules to increase the inhibitory activity of MMP-9 as an anticancer agent. Based on the pharmacophore, amide cinnamate derivatives were selected for the design of newer analogs by forming amide linkages with alkyl and aryl. Among several amide cinnamate derivatives, phenyl amide cinnamate (8), methoxyphenyl amide cinnamate (9), and octyl amide cinnamate (7) were found to be closer and interact better at the MMP-9 protein-binding site. The amide cinnamate compound exhibited a binding pattern almost similar to that of the native 4 WZV receptor ligand. The amino acid residues important in MMP-9 protein-binding were Leu188, Glu277, His226, Tyr248, Arg249, Met247, Tyr245, Leu243, and Ala242. The amide cinnamate compound represented an initial breakthrough in the design of several effective MMP-9 inhibitors for potential cancer treatment. The molecular docking results indicated that phenyl amide cinnamate (8), methoxyphenyl amide cinnamate (9), and octyl amide cinnamate (7) were thought to act as strong inhibitors of MMP-9. The visualization of the interaction of the ligand-binding mode with the MMP-9 protein is shown in Figure 3. The results of molecular docking revealed that there is a notable correlation between the binding mode of the ligand and the amino acid residues, which is consistent with the findings from the in vitro bioactivity test. Additionally, the in silico study of docking amide cinnamate compounds with a cancer receptor protein showed a significant correlation with the in vitro study of amide cinnamate compounds on MCF-7 cancer cells. This correlation is evident in Figure 4, where there is a significant correlation between the predicted and experimental ΔG values.
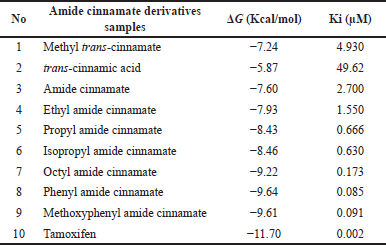 | Table 2. Molecular docking of amide cinnamate derivative results. [Click here to view] |
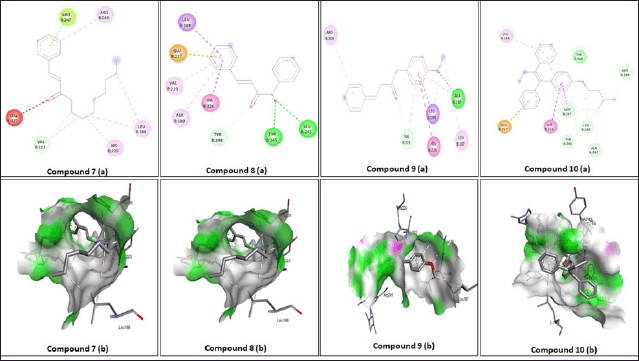 | Figure 3. (a) 2D and (b) 3D interaction of binding mode of amide cinnamate derivatives with 4WZV enzyme. [Click here to view] |
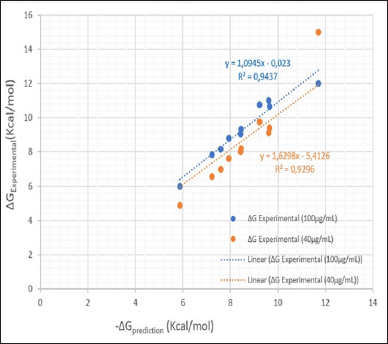 | Figure 4. Correlation of ΔGprediction and ΔGexperimental of amide cinnamate derivatives at sample concentration of 100 and 40 μg/ml. [Click here to view] |
CONCLUSION
To summarize, this study identified three amide cinnamate derivatives, phenyl amide cinnamate (8), methoxyphenyl amide cinnamate (9), and octyl amide cinnamate (7) with potent inhibitory activity against MCF-7 cells. The presence of amide substituents of cinnamate led to altered MCF-7 cells’ inhibitory activity, which was supported by the docking results showing a significant correlation between the ligand binding mode and amino acid residues observed in the in vitro bioactivity test. Among the six amide cinnamate derivatives, phenyl amide cinnamate (8) exhibited the highest potent inhibitory activity (94.8% and 90.8% inhibition at 100 and 40 μg/ml, respectively) and also showed the best activity of interaction with MMP-9 protein in the molecular docking results (ΔG = −9.64 Kcal/mol, Ki = 0.085 µM). Therefore, further evaluation of the activity of this compound is warranted.
ACKNOWLEDGMENTS
We are very grateful for the funding that was given by Research and Innovation for Indonesia advanced to wave 2 (RIIM stage 2)—National Research and Innovation Agency (BRIN) No 82/II.7/HK/2022. In addition, we are also very thankful for all the support and help from the Research Center for Pharmaceutical Ingredients and Traditional Medicine and Research Center for Chemistry—National Research and Innovation Agency of Indonesia (BRIN). We are also very thankful to Dr. Churiah for the Breast cancer cell line MCF-7. TE and NA contributed equally to this work as main contributors.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Welch DR, Harper DE, Yohem KH. U-77,863: a novel cinnamide isolated from Streptomyces griseoluteus that inhibits cancer invasion and metastasis. Clin Exp Mestastasis. 1993;11:201–12. CrossRef
2. Ernawati T, Nurhalimah N, Minarti M. Sintesis N-Oktilsinamamid dan Aktivitasnya terhadap Sitotoksik Sel Kanker Leukemia P388. J Kim Valensi. 2017;3(2):128–34. CrossRef
3. Li QF, Shi SL, Liu QR, Tang J, Song J, Liang Y. Anticancer effects of ginsenoside Rg1, cinnamic acid, and tanshinone in osteosarcoma MG-63 cells: downregulation and cytoplasmic trafficking of neucleophosmin. Int J Biochem Cell B. 2008;40:1918–29. CrossRef
4. Ernawati T, Fairusi D. Sintesis fenil sinamat dan 4-fenilkroman-2-on dan Uji Sitotoksisitas terhadap sel kanker serviks HeLa. J Ilmu Kefarmasian Indones. 2013;11(2):202–10. Available from: http://jifi.farmasi.univpancasila.ac.id/index.php/jifi /article/view/221.
5. Akao Y, Maruyama H, Matsumoto K, Ohguchi K, Nishizawa K, Sakamoto T, et al. Cell growth inhibitory effect of cinnamic acid derivatives from propolis on human tumor cell lines. Bio Pharm Bull. 2003;26(7):1057–9. CrossRef
6. Banskota AH, Nagaoka T, Sumioka LY, Tezuka Y, Awale S, Midorikawa K, et al. Antiproliferative activity of the Netherlands propolis and its active principles in cancer cell lines. J Ethnopharmacol. 2002;80:67–73. CrossRef
7. Hasegawa T, Bai J, Dai J, Bai L, Sakai J, Nishizawa S, et al. Synthesis and structure-activity relationships of taxuyunnanine C derivatives as multidrug resistance modulator in MDR cancer cells. Bioorg Med Chem Lett. 2007;17:3722–8. CrossRef
8. Atsumi T, Sanjiki T, Kiyohara T. Imidazole derivatives, their preparation and their pharmaceutical compositions. European Patent EP. 0075629. 1983. Available from: https://patents.google.com/patent/EP0194984A1/en
9. Cozzi P, Beria I, Biasoli G, Caldarelli M, Capolongo L, Geroni C, et al. Novel phenyl nitrogen mustard and half-mustard derivatives of distamycin A. Bioorg Med Chem Lett. 1997;7:2985–90. CrossRef
10. Boger DL, Johnson DS, Yun W. (+)- and ent-(-)- Duocarmycin SA and (+)- and ent-(-)-NBoc- DSA DNA alkylation properties. Alkylation site models that accommodate the offset AT-rich adenine N3 alkylation selectivity of the enantiomeric agents. Am Chem Soc. 1994;116:1635–56. CrossRef
11. Pardin C, Roy I, Chica RA, Bonneil E, Thibault P, Lubell WD, et al. Photolabeling of tissue transglutaminase reveals the binding mode of potent cinnamoyl inhibitors. Biochemistry.2009;48:3346–53. CrossRef
12. Levistzki A, Willingham M, Pastan IH. Evidence for participation of transglutaminase in receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1980;77:2706–10. CrossRef
13. Chang S, Yin SL, Wang J, Jing YK, Dong JH. Design and synthesis of novel 2-phenylaminopyrimidine (PAP) derivatives and their antiproliferative effects in human chronic myeloid leukemia cells. Molecules. 2009;14:4166–79. CrossRef
14. Srivastava V, Srivastava AM, Tiwari AK, Srivastava R, Sharma R, Sharma H, et al. Disubstituted 4(3H) quinazolones: a novel class of antitumor agents. Chem Biol Drug. 2009;74:297–301. CrossRef
15. Liu Y, Shang L, Fang H, Zhu H, Mua J, Wang Q, et al. Design, synthesis, and preliminary studies of the activity of novel derivatives of N-cinnamoyl-l-aspartic acid as inhibitors of aminopeptidase N/CD13. Bioorg Med Chem. 2009;17:7398–404. CrossRef
16. De P, Baltas M, Lamoral-Theys D, Bruyere C, Kiss R, Bedos-Belval F, et al. Synthesis and anticancer activity evaluation of 2(4-alkoxy phenyl) cyclo propyl hydrazides and triazolo phthala zines. Bioorg Med Chem Lett. 2010;18:2537–48. CrossRef
17. Guo Z. The modification of natural products for medical use. Acta Pharm Sin B. 2017;7:119–36. CrossRef
18. Mannello F, Tonti G, Papa S. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr Cancer Drug Targets. 2005;5:285–98. CrossRef
19. Shi ZH, Li NG, Shi QP, Tang Y, Tang YP, Li W, et al. Synthesis and structure-activity relationship analysis of caffeic acid amides as selective matrix metallo proteinase inhibitors. Bioorg Med Chem Lett. 2013;23:1206–11. CrossRef
20. Paramvir S, Ajmer S, Grewal DP, Viney L. Synthesis and evaluation of a series of caffeic acid derivatives as anticancer agents. Future J Pharm Sci. 2018;4:124–30. CrossRef
21. Ernawati T. Penapisan virtual senyawa turunan metil sinamat terhadap enzim siklooksigenase-2 (COX-2). J Kim Terap Indones. 2012;14(2):66–70. CrossRef
22. Ernawati T, Anita Y, Lotulung PD, Hanafi M. Synthesis of methyl 2-cinnamamido-3-hydroxy propanoate having activity against P388 leukemia cells. J App Pharm Sci. 2014;4(3):92–5. CrossRef
23. Ernawati T, Muním A, Hanafi M, Yanuar A. Synthesis of cinnanamide derivatives and their α-glucosidase inhibitory activities. Sains Malays. 2020;49(2):93–100. CrossRef
24. Chauffert B, Pelletier H, Corda C, Solary E, Bedenne L, Caillot D, et al. Potential usefulness of quinine to circumvent the anthracycline resistance in clinical practice. Br J Cancer. 1990;62(3):395–7. CrossRef
25. Solary E, Mannone L, Moreau D, Caillot D, Casasnovas RO, Guy H, et al. Phase I study of cinchonine, a multidrug resistance reversing agent, combined with the CHVP regimen in relapsed and refractory lymphoproliferative syndromes. Leukemia. 2000;14:2085–94. CrossRef
26. Dewanjee S, Dua TK, Bhattacharjee N, Das A, Gangopadhyay M, Khanra R, et al. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules. 2017;22(6):871. CrossRef
27. Liu L, Ju R, Xie H, Zeng F, Zhang C, Zhao W, et al. Efficacy and mechanism of a compound epirubicin plus quinine injection for the treatment of drug-resistant breast cancer. J Chinese Pharm Sci. 2015;24(9):563–71. CrossRef
28. Yanuar A. Penambatan molekule.1st ed. Depok, Indonesia: Fakultas Farmasi Universitas; 2012.
29. Seo WD, Kim JH, Kang JE, Ryu HW, Curtis-Long MJ, Lee HS, et al. Sulfonamide chalcone as a new class of α-glucosidase inhibitors. Bioorg Med Chem Lett. 2005;15:5514–20. CrossRef