
INTRODUCTION
Therapeutic drug monitoring (TDM) is one of the clinical pharmacy services in hospitals [1], which is essential for a drug with a narrow therapeutic index, pharmacokinetic (PK) variability, and severe toxicity or side effects [2]. Amikacin (AMK) is an aminoglycoside (AMG) antibiotic commonly used to treat serious infections such as sepsis, septic shock, and ventilator-associated pneumonia [3–7]. AMG’s nephrotoxicity and ototoxicity may be related to its high peak concentration (Cmax) or trough concentration (Cmin) [8,9], especially in patients with renal impairment [10]. Because AMK is a concentration-dependent antibiotic with a narrow therapeutic index and dose-related toxicity, monitoring AMK blood levels is critical to ensure the safe and effective administration of AMK. Unfortunately, TDM in Indonesian hospitals has not been widely implemented due to various limitations, including costs, equipment, and human resources.
Determination of drug levels can be done by various analytical methods, depending on the drug’s physicochemical properties, drug concentrations, costs, available analytical instruments, and the amount and nature of the samples [11]. Immunoassays are easy to adopt but lack specificity and accuracy, have limited ability to analyze drugs, and incur a high cost per sample [12]. AMK and other AMGs are highly polar and have no natural chromophore, which is a significant challenge in analyzing AMG directly by high performance liquid chromatography (HPLC) [13], thus requiring a chemical derivatization process [14]. A method suitable for determining AMK levels in human plasma is liquid chromatography-tandem mass spectrometry (LC-MS/MS) [13–16]. LC-MS/MS is an essential instrument in TDM because it has higher sensitivity and specificity than other methods and can quantify compounds without natural chromophores or fluorophores [10]. It can measure highly polar analytes without a derivatization process, providing more flexibility because it can be developed independently, produces qualitative and quantitative data [17], allows for a limited sample volume, and has a lower cost per sample as it can quantify several antibiotics simultaneously [15]. The main disadvantages of LC-MS/MS are the relatively high cost of the instrument and increased assay complexity [10]. However, the ability of the MS detector to specifically identify the target analytes based on their masses makes the optimization of LC conditions for separation in complex matrices much faster than conventional LC.
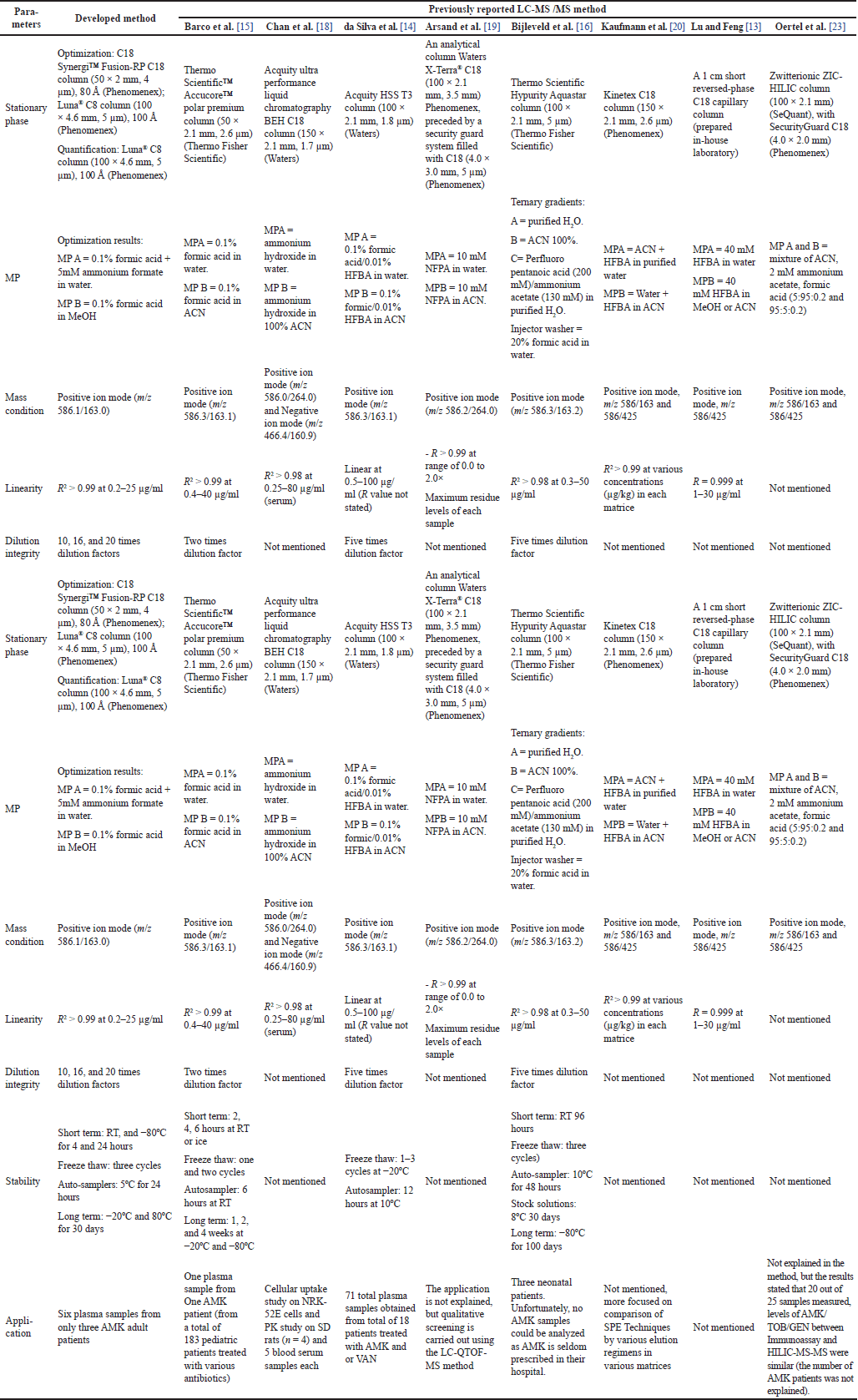
Previous studies on AMK determination by the LC-MS/MS method used various types of columns and mobile phases (MP) [13–16,18,19]. Sample preparation strategies for the extraction of AMK and other AMGs samples also vary, generally using solid-phase extraction (SPE) [18,20–23] or the protein precipitation (PP) method, which uses various deproteinizing agents such as heptafluorobutyric acid (HFBA) [13], trichloroacetic acid (TCA) [16], TCA and methanol (MeOH) [15], or acetonitrile [14,18]. However, to the best of our knowledge, a PP method using MeOH alone as a solvent in sample preparation for the AMK analysis in human plasma has not been previously reported. The PP method developed in this new method uses only MeOH as a solvent and can be performed quickly and easily. MeOH is also used as a stock solution solvent, organic MP, and injector wash, making it more efficient in chemical usage. In addition, this research also conducted MP and gradient optimization, as well as column optimization (C18 and C8), compared to previous studies, which mostly used C18 columns [13–16,18–20] or hydrophilic interaction chromatography (HILIC) columns [23].
This research will focus on developing and validating an efficient LC-MS/MS method for determining AMK levels in human plasma, with a PP technique using MeOH as a sample extraction method. MeOH is inexpensive and commonly used solvent in the laboratory. The goal is to obtain a simple, fast, and easy procedure with minimal costs, which can be applied in laboratories in Indonesia and other countries with the same constraints related to the implementation of TDM. The method will be used on adult inpatients with AMK therapy at Dr. Sardjito Central General Hospital in Yogyakarta Special Region Province, Indonesia.
MATERIALS AND METHODS
Reagents and chemicals
AMK (LRAC9136) was purchased from Sigma-Aldrich (Milan, Italy). Gentamicin sulfate (GS) (B0316ISB, BPFI) as an internal standard (IS) was obtained from the Indonesian National Agency for Drug and Food Control (BPOM RI, Jakarta, Indonesia). MeOH (gradient grade), formic acid (MS grade), and ammonium formate (MS grade) were obtained from Sigma-Aldrich (Milan, Italy). Water for injection was purchased from Ikapharmindo Putramas (Jakarta, Indonesia). In addition, drug-free human plasma samples were obtained from healthy volunteers at the Indonesian Red Cross, Yogyakarta, and clinical plasma samples from adult hospitalized patients at Dr. Sardjito General Hospital, Yogyakarta, Indonesia.
Instrumentations and LC-MS/MS conditions
Experiments were carried out at the Advanced Pharmaceutical Sciences Laboratory, Faculty of Pharmacy, Universitas Gadjah Mada (Yogyakarta, Indonesia) using LC-MS/MS (Exion LC system (Sciex, Singapore) combined with a Sciex Triple-Quadrapole 4500 Mass Spectrometer with positive ion multiple reaction monitoring (MRM) mode, ion source CUR 35.0, CAD 7, IonSpray voltage (IS) 5500 V, and temperature (TEM) 500ºC. Two kinds of columns used in the optimization were Synergi™ 4 µm Fusion-RP C18 80 Å, 50 × 2 mm LC column (Phenomenex Inc, California, US) and Luna® 5 µm C8 100 Å, 100 × 4.6 mm LC column (Phenomenex Inc, California, US). The latest was chosen for the method validation and AMK quantification. The MP used for AMK quantification was obtained based on the optimization of the method, which consisted of an aqueous mobile phase A (MP A) composed of 0.1% formic acid + 5 mM ammonium formate in water and an organic mobile phase B (MP B) consisting of 0.1% formic acid in MeOH, with a gradient of MP A:B (0–2 minutes: 95:5, 2–3 minutes: 75:25, 3–4.5 minutes: 5:95, 4.5–7.5 minutes: 95:5), with the total run time of 7.5 minutes.
Analytical procedures
Preparation of calibration standard and quality control (QCs) samples
A series of AMK standard solutions (4–500 µg/ml) and IS solution (100 µg/ml) were prepared in MeOH. The standard calibration series were made by spiking the AMK standard solutions and IS solution into the plasma to obtain 0.2–25 µg/ml of AMK and 10 µg/ml of IS as the final concentration. In addition, AMK QC solutions were prepared in 0.2 µg/ml [lowest limit of quantification (LLOQ)], 0.6 µg/ml (Low QC), 15 µg/ml (Medium QC), and 20 µg/ml (High QC).
Optimization of LC-MS/MS condition and PP method
The optimization was carried out to the column, MP, MP gradient, injection volume, and flow rate. Method optimization was initially conducted based on previous research [15]. LC analysis was initially performed using a reversed-phase system with a Synergi™ 4 µm Fusion-RP C18 80 Å column. The initial MPs were MP A (0.1% formic acid in H2O) and MP B (0.1% formic acid in MeOH), using a gradient elution technique A:B (0–3 minutes: 80:20, 3–4.5 minutes: 10:90, 4.5–7.5 minutes: 80:20), with 0.2 ml/minute initial flow rate, and the injection volume was 10 µl.
The optimization of sample preparation using the PP (deproteinization) process was carried out using MeOH with an optimum composition ratio based on the recovery of test samples. The ratio of plasma: MeOH for PP was 1:3 up to 1:9. Into each tube of AMK spiked plasma solutions, the IS solution (100 µg/ml) and MeOH were added with the optimal volume obtained in the PP optimization process. The solution was centrifuged for 10 minutes at 12,000 rpm at 4ºC. A total of 500 µl of supernatant was put into a new tube. MP A (according to the optimization results) was added, homogenized, and filtered using a 0.45 µm nylon filter membrane, then injected into the LC-MS/MS system.
Method validation
Validation was carried out based on the Guideline on Bioanalytical Method Validation of the European Medicines Agency (EMA) EMA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2 [24] and the Methodological Guidelines for Specific Bioequivalence Tests of Active Substances of the Indonesian National Agency for Drug and Food Control (BPOM RI) [25]. Analytical method validation parameters include selectivity, carry-over, LLOQ, calibration curve linearity, accuracy, precision, dilution integrity, and stability [24]. In addition, the LC-MS/MS system conditions and PP process for sample preparation in all method validation procedures were performed based on the optimization results obtained.
Linearity of calibration curve
Linearity was evaluated as the ratio of analyte peak area versus theoretical concentration in three sets of curves in a minimum of six concentrations, each run on three different days. Calibration standards met the criteria if the difference in concentration of the measured standard solution with the theoretical concentration was not more than ±15%, except for LLOQ, which was not more than ±20%. In addition, at least 75% of the concentration of the standard solution must fulfill the criteria [24]. For each calibration curve, linear regression analysis also determined the slope, intercept, and correlation coefficient [15].
Lowest limit of quantification
The LLOQ determination was performed by injecting AMK spiked plasma at 0.1, 0.2, and 0.3 µg/ml, each with at least five replicates. The LLOQ was calculated at measured concentrations with a 20% deviation (accuracy ranged from 80% to 120% compared to theoretical concentrations).
Selectivity and carry-over
Selectivity tests were carried out by injecting six blank plasma to be analyzed and evaluated for interference to AMK peaks. Blank plasma was prepared as directed in the sample preparation without adding the AMK and IS solution. Meanwhile, blank plasma injection was carried out after the injection of AMK standard calibration solution at the highest concentration in the carry-over test. The analytical method met the selectivity criteria if the peak interference at the AMK retention time (RT) in blank plasma was less than 20% of the area at LLOQ AMK. Carry-over obtained in the blank sample after injecting high concentration standard should not exceed 20% of the LLOQ [24].
Accuracy and precision
Accuracy and precision were evaluated in QCs samples (LLOQ, Low QC, Medium QC, and High QC). Measurements were performed with five replicates, each run on three different days. The accuracy test met the criteria if the average measured concentration is within 15% of the nominal values, except for the LLOQ, which must not be more than 20%. Meanwhile, the method meets the precision test criteria if the relative standard deviation (RSD) or the coefficient of variation of the QC sample concentration measured with five replicates does not exceed 15%, except for LLOQ, which should be within 20% [24].
Dilution integrity
Dilution integrity was performed by spiking blank plasma with AMK concentrations above the upper limit of quantification, then diluting with blank plasma (with a dilution factor of 10–20 times) at least five replicates per dilution factor. Accuracy and precision should be met the required criteria, which is within ±15% [24].
Stability tests
AMK stability tests were conducted using low and high QC samples that were analyzed immediately after preparation and samples stored under certain storage conditions, each with three replicates. The stability tests performed include short-term stability, which was carried out on low and high QC samples that were immediately prepared at room temperature, frozen in a freezer (−80ºC) for 4 hours (T4), and 24 hours (T24), and then thawed at room temperature. In addition, freeze-thaw stability was tested on AMK samples stored in a freezer (−80ºC) and then thawed at room temperature with three freeze-thaw cycles. Autosampler stability was performed on AMK-processed samples ready to be injected and stored in an autosampler rack 24 hours before injection. Each stability test meets the criteria if the average concentration at each level is within ±15% of the nominal value [24].
Assay application on clinical samples
The optimized and validated LC-MS/MS method was then applied to measuring AMK blood levels in hospitalized adult patients. The application phase of the LC-MS/MS method for AMK quantification was conducted at Dr. Sardjito Central General Hospital, Yogyakarta, Indonesia. This study has received ethical approval from the Health Research Ethics Committee of the Faculty of Medicine, Universitas Gadjah Mada, Yogyakarta, with the number KE/FK/0885/EC/2021. Data collection in the hospital was carried out prospectively using the purposive sampling method with a limited number of research subjects (n = 3). The study subjects were adult hospitalized patients at Dr. Sardjito Hospital from July to September 2022. The inclusion criteria were patients aged >18 years, diagnosed with infectious disease, receiving AMK therapy for at least 3 days with intermittent IV infusion routes, and signing informed consent forms to become research subjects. The exclusion criteria were patients with tuberculosis, patients with immunocompromised or hematologic malignancies, or patients who died before the third day of AMK therapy. The study began with patient screening, recruitment, and informed consent to the patient or the patient’s family, and permission from the patient’s doctor. To determine AMK plasma levels, blood samples were taken during a steady state (within 48th to 96th hours after the first dose of AMK). Sampling was done at least twice at 3 ml: before AMK dosing to measure the trough concentration (Cmin) and 30 or 60 minutes after AMK infusion started to measure peak concentration (Cmax). Blood samples were collected in K3-EDTA tubes, then immediately prepared into plasma at the hospital’s Integrated Laboratory Installation and stored at −80ºC until the quantification.
RESULTS
Methods optimization
The LC-MS/MS method was developed for AMK quantification in human plasma by optimizing LC-MS/MS conditions and the PP method. Table 1 presents the MRM ion transitions used in generating this method. For the subsequent AMK quantification process, MRM ion transitions were used, including 586.136/163.00 for AMK and 478.136/322.100 for GS as the IS. Based on the method optimization, it was obtained that the optimal LC-MS/MS conditions for determining AMK levels in plasma can be performed using a C8 column, MP (MP A: 0.1% formic acid + 5 mM formic ammonium in water; MP B: 0.1% formic acid in MeOH), MP A:B gradient (0–2 minutes: 95:5, 2–3 minutes: 75:25, 3–4.5 minutes: 5:95, 4.5–7.5 minutes: 95:5), a flow rate of 0.5 ml/minute, injection volume of 5 µl, RT within 1.4 minutes, with a total run time of 7.5 minutes. In addition, MeOH with the optimal final total plasma: MeOH ratio of 1:9 was also proven to be used in the AMK extraction process using a PP method, which was relatively simple and fast. In this case, 100 µl of plasma was spiked with 100 µl of AMK standard, added with 100 µl of IS, and 700 µl of MeOH. After centrifuging, the supernatant was added with MP A, homogenized, filtered, and injected into the LC-MS/MS system. These optimal conditions produced AMK and IS chromatograms with good peak shapes and symmetry, showing AMK % recoveries within the acceptable 85%–115% range.
Chromatography
The chromatogram obtained in the AMK quantification in plasma using the LC-MS/MS method showed a symmetrical and sharp peak shape with a baseline resolution of fewer than 10 seconds at a RT of 1.4 minutes. Figure 1 illustrates LC-MS/MS chromatogram representation for drug-free plasma, AMK, IS spiked plasma, and clinical samples.
Method validation
Table 2 depicts the results of the method validation in this study. The AMK response was linear at 0.2–25 µg/ml concentrations, with the R2 value of the standard calibration curve on all curve sets reaching >0.99. This method has also fulfilled the standard calibration criteria since each minimum of six standard concentrations for 3 days produced % of differentiation in the range of 0.0%–13.8%. The LLOQ test data showed that the percentage of the difference between the measured and theoretical concentrations is <20% or a recovery value in the range of 80%–120% was produced at a concentration of 0.2 µg/ml. Thus, LLOQ was determined at a concentration of 0.2 µg/ml.
The developed method showed excellent selectivity, indicated by the absence of interfering peaks at the AMK peak RT from the blank plasma injection. The results of the carry-over test also depicted no AMK peak interference (0.0%) in the blank plasma following the injection of an AMK standard solution at the highest tested concentration. As a result, the QCs samples’ accuracy was within the acceptable range, i.e., from 93.81%–110.04%. Meanwhile, RSD values from the within-run and between-run precision were <15% (ranging from 2.3% to 9.5%), except for the LLOQ samples that ranged from 5.6% to 18.9% (required RSD for LLOQ is <20%). The dilution integrity testing using 1:10, 1:16, and 1:20 diluted plasma resulted in 104.7%–108.5% recovery and 2.2%–3.6% RSD values. Thus, based on the results of the dilution integrity testing, this analytical method has also met the criteria for accuracy and precision. Furthermore, the stability test showed that the analytical method developed met the criteria for good sample stability. Recovery percentages from the short-term stability test at room temperature and −80ºC for 4 and 24 hours, the freeze-thaw stability test, the stability test for 24 hours in an autosampler, and long-term stability at −20ºC and −80ºC for 30 days were within the acceptable range of 92.2%–108.2%, 90.8%–108.6%, 87.2%–97.3%, and 85.1%–105.4%, respectively. Table 3 presents the complete stability test results.
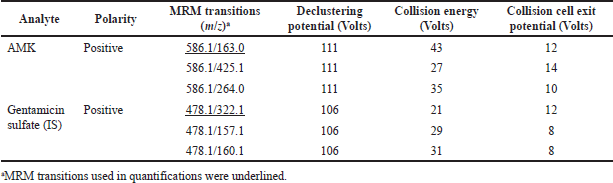 | Table 1. MS ionization conditions in the development of LC-MS/MS method for the determination of AMK in human plasma. [Click here to view] |
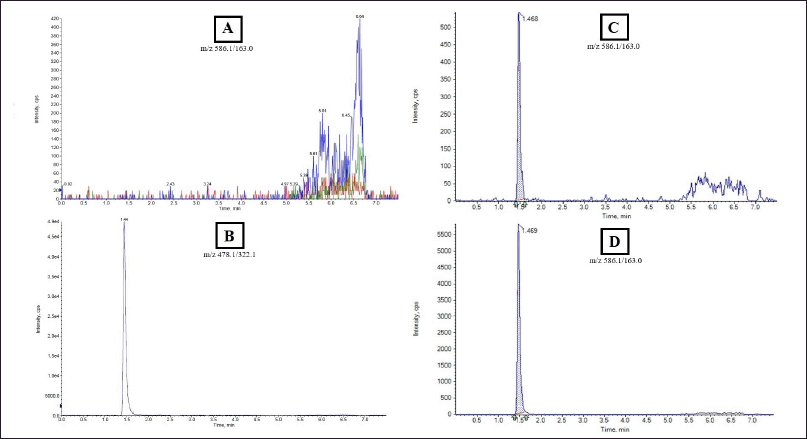 | Figure 1. LC-MS/MS chromatograms obtained from human plasma analysis: (A) drug-free plasma; (B) blank plasma spiked with IS (10 μg/ml); (C) sample at low QC (0.6 μg/ml); (D) a random plasma sample of a patient receiving AMK. [Click here to view] |
Assay application
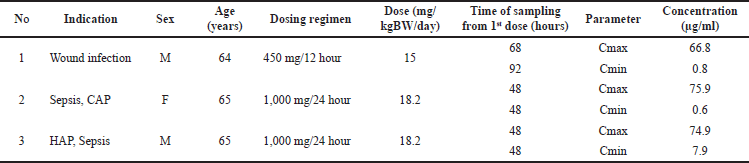
The developed LC-MS/MS method has been tested on clinical samples to measure AMK levels in plasma samples from three adult inpatients at Dr. Sardjito Central Public Hospital Yogyakarta, Indonesia. The results are presented in Table 4. The research subjects comprised two men and one woman aged 64 to 65 with various indications and AMK dosage regimens. AMK Cmax in patient plasma obtained from measurement results was 66.8 to 75.9 µg/ml. Meanwhile, the AMK Cmin of the three subjects were measured between 0.6 and 7.9 µg/ml.
DISCUSSION
This research has succeeded in developing an LC-MS/MS method for analyzing AMK levels in human plasma that is relatively simple, short, and uses chemicals commonly found in laboratories. Method development was done by optimizing LC-MS/MS conditions and sample extraction methods by quantifying product ions at m/z 163.0 for AMK and 322.1 for IS (Table 1) to produce the peaks with the best intensity. The development was started using a Synergy™ Fusion-RP C18 column with 0.1% formic acid in water (MP A) and MeOH (MP B) as an eluent. The MP was chosen based on previous research [15], but in this present study, acetonitrile in MP B was replaced by MeOH. Previous studies showed good separation when MeOH or acetonitrile was used as eluent with gradient chromatography [13].
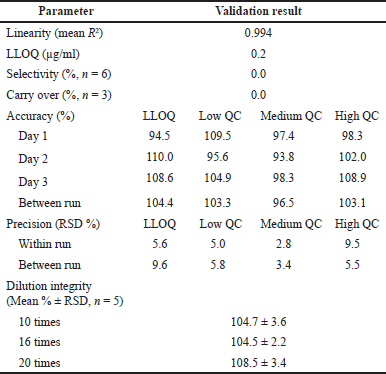 | Table 2. Validation results of the LC-MS/MS method for the determination of AMK in human plasma. [Click here to view] |
In the initial condition, AMK and IS peak shapes were not good and eluted very early at a RT of 0.52 minutes (Supplementary Fig. 1), not far different from the results of other studies at an RT of 0.48 minutes [15]. Because of its polarity, AMK eluted quickly by the MP at the beginning was a major challenge for AMG separation with chromatography [18] because it tended to overlap with the components in the matrix. In addition, AMK and other AMG are polar molecules and not retained in the reversed-phase column, so it needs ion-pair reagents to improve their RT, such as HFBA [13]. However, HFBA might contaminate the MS systems and was not ideal for testing other compounds, particularly those run in negative mode. pH adjustment could be made to maintain the unionized form of AMG, such as using ammonium hydroxide [18]. In this study, we adjusted pH using 0.01 M NaHCO3 in MP A, but AMK and IS peaks did not appear.
Another alternative to increase AMK RT was replacing the column with a C8 or HILIC column. However, HILIC columns are known to be less stable, as they are mainly determined by the ionic strength and pH of the MP [14,26]. Hence, we changed the column to C8 using the initial MP composition to obtain a good peak. This C8 column could produce AMK peaks in the RT range of 2.4–3.3 minutes, but a broad peak shape of the IS was obtained (Supplementary Fig. 1). Ammonium formate was added to the MP A, with the initial MP A gradient of 95%, then decreasing until 4.5 minutes and increasing again in 4.5–7.5 minutes, the flow rate of 0.5 ml/minute, and an injection volume of 5 µl. With these changes, sharp and symmetrical AMK and IS peaks could be produced at RT of 1.4 minutes. Thus, it can be concluded that the optimal LC-MS/MS condition for AMK quantification in plasma in this study was achieved by using a C8 column, an aqueous MP of 0.1% formic acid + 5 mM ammonium formate in water, an organic MP of 0.1% formic acid in MeOH, with a total run time of 7.5 minutes. This simpler and more affordable method produced the same result as some previous studies that used various columns and MP with a total run time in the range of 5.0 to 10 minutes [13–16,18]. Compared with HPLC analysis, the shorter run time of the LC-MS/MS method has several advantages, including improved productivity, fast turnaround time, and efficient use in MP [15].
The previous AMK analysis studies commonly carried out sample preparation using the SPE technique [18,20–23,27,28]. However, this method is less effective for general use, as the process is more complicated, time-consuming, and costly [19]. Another simpler and widely used method is the PP or deproteination technique using various deproteinating agents [13–16,18]. Organic solvents such as MeOH, ethanol, and acetonitrile can precipitate proteins. This precipitation was related to the isoelectric point of the protein, where the farther the isoelectric point, the more increasing the solubility. Adding organic solvent decreased the solvent’s or water’s dielectric constant, thereby increasing the tension between charged molecules and facilitating protein electrostatic interactions. Organic solvents might also replace some water molecules around the hydrophobic areas of the protein surface, reducing the water concentration. As a result, protein solubility will decrease, and precipitation will occur [17]. In this study, PP using MeOH was chosen because MeOH is easy to find, very commonly used in laboratories, and relatively affordable to minimize the analysis cost. This study also used MeOH as an organic MP (MP B). The optimal volume of MeOH used in the PP process was 900 per 100 µl plasma with a recovery percentage in QCs samples that met the requirements of 85%–115% and chromatograms that remained in good shape and resolution at a relatively similar RTof 1.4 minutes.
Analytical methods must be validated to meet the required parameters [29]. Validation of the LC-MS/MS method was necessary to ensure the reliability of the measured AMK concentrations. The developed LC-MS/MS method for AMK quantification has also been validated and met the requirements for all tested parameters based on the EMA Guidelines [24] (Table 2). In this study, the LLOQ value obtained was 0.2 µg/ml, relatively low and in accordance with the results of previous studies determining LLOQ AMK in the range of 0.125–0.500 µg/ml [14–16,18,19]. Thus, it is expected that it will be able to measure the lowest levels (Cmin) of AMK in the plasma of patients. This low LLOQ value is one of the advantages of the LC-MS/MS method compared to other methods, such as immunoassay, often clinically used in determining blood drug levels. In addition, immunoassay, in general, has relatively large LLOQ values, i.e., in the range of 1–3 mg/l, as well as the limitations of antibodies that can bind to other molecules such as drug metabolites or other biological molecules that are present in the sample matrices [18].
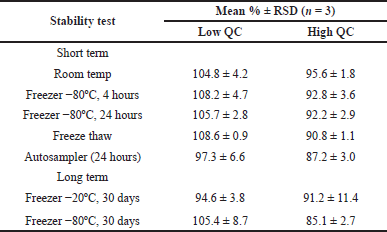 | Table 3. Summary of various stability results. [Click here to view] |
 | Table 4. Results of the measurement of AMK from clinical samples. [Click here to view] |
 | Table 5. Comparison of performance between the proposed method and published LC-MS/MS methods. [Click here to view] |
This method produced excellent accuracy and precision on the same day (within-run) and different days (between-run), all within the required and acceptable range. Similarly, the dilution integrity test results with a dilution factor of 10×, 16×, and 20× showed acceptable accuracy and precision (Table 2). A good accuracy level in up to 20 times dilution makes this method possible to measure AMK up to very high levels. Based on the stability test results (Table 3), the samples can be stored in various TEM in the short term or autosampler and still showed good stability up to three cycles of freeze-thaw conditions. Based on the long-term stability testing, the AMK sample may also be stored at −20ºC or −80ºC for 30 days after sampling. Various previous studies using the LC-MS/MS method also obtained that AMK sample stability was satisfactory and met the validity requirements, both in short-term stability tests, freeze-thaw, and autosampler [14–16], and long-term stability tests for 1–4 weeks at −20ºC and −80ºC storage [15], and 30 days at 80ºC and 100 days at −80ºC [16]. Thus, the LC-MS/MS allows measuring AMK levels in samples stored for quite a long time with a good recovery.
Assay application in clinical samples was carried out on six blood plasma samples collected from three adult hospitalized patients receiving AMK therapy at a dosing regimen of 450 mg/12 hours or 1 g/24 hours. Each patient’s blood sample was taken at different times, 48–92 hours from the first AMK given, to obtain Cmax and Cmin levels in steady state condition. The method that has been developed can measure Cmax and Cmin from the three subjects with varying concentration ranges (Table 4). The measured Cmin were all still above LLOQ, but Cmax was higher than the linear calibration concentration. In the case of concentrations higher than the linear range, the sample can be diluted up to 20 times with good accuracy and precision based on the results of the dilution integrity test (Table 2). The minimum AMK levels obtained in two patients were relatively low (<1 mg/l), probably as a consequence of clearance of AMK [14], where the AMK t1/2 elimination was around 2 hours (range 1.4–2.3 hours) in patients with normal kidneys [8]. Therefore, AMK is likely to have been almost eliminated in about 10–14 hours. Some previous research also tested the application of the LC-MS/MS method to clinical plasma samples in one AMK patient with a measured Cmin of 5.1 mg/l [15], three neonatal patients [26], and 18 patients with levels obtained were 0.5–113.6 mg/l and one sample below the LLOQ [14].
Overall, this study successfully developed and validated a method for determining AMK levels in human blood plasma by LC-MS/MS that is efficient and simple without complex protocols. It was also successfully applied to patients’ samples in the hospital. Compared to previous LC-MS/MS methods for determining AMK in various matrices (Table 5), this study showed that the C8 column could be used in the determination of highly polar drugs such as AMK, using the simple PP procedures, and allows for the efficient use of MeOH as a stock solvent, deproteinizing agent, MP B, and injector washer. However, the limitations of this study include the relatively small number of clinical samples used (a total of six plasma samples obtained from three subjects). Therefore, further research is needed to evaluate the application of this method to a more significant number of subjects and conducted in different regions.
Nevertheless, this LC-MS/MS method was expected to be widely used in hospitals, especially in TDM activities, to obtain optimal AMK therapy for patients. However, another study limitation was the sole determination of AMK (with GS as IS), compared to other published methods reporting the quantification of some antibiotics simultaneously. Thus, further studies need to be conducted to evaluate the ability of this LC-MS/MS method to determine the levels of other antibiotics simultaneously, especially those typically used in conjunction with therapy. This simultaneous determination may provide a highly effective assessment of PK pharmacodynamic parameters and applying TDM antibiotics clinically because sometimes AMK was given in combination with other antibiotics.
CONCLUSION
The LC-MS/MS method has been developed for simple and efficient analysis of AMK levels in plasma. The technique has fulfilled all validation requirements and has been successfully applied to clinical samples in hospitalized patients. However, further research is needed with more patient samples to evaluate the impact of AMK plasma levels on clinical outcomes and toxicity.
ACKNOWLEDGMENT
The authors would like to thank the Ministry of Education and Culture Republic of Indonesia through the 2022 Doctoral Dissertation Research (PDD 2022) grant, Pertamina through the 2021 Corporate Social Responsibility fund, and the Faculty of Pharmacy UGM through the 2022 Faculty of Pharmacy grant for providing the funding for this researchand Dr. Intan Pristian Yuliani and Setiani Silvy for the assistance in data collection processes in the hospital.
AUTHOR CONTRIBUTIONS
All authors have substantial contributions to the conception and design of the work, acquisition of data, or analysis and interpretation of data for this research; took part in drafting the manuscript or revising it critically for important intellectual content; agreed to submit to this journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. Therefore, all the authors are eligible to be authors per the International Committee of Medical Journal Editors (ICMJE) requirements.
CONFLICTS OF INTEREST
All authors report no financial or other conflicts of interest in this research.
ETHICAL APPROVALS
This study has received ethical approval from the Health Research Ethics Committee of the Faculty of Medicine, Universitas Gadjah Mada, Yogyakarta, with the number KE/FK/0885/EC/2021.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Menteri Kesehatan RI. Standar Pelayanan Kefarmasian di Rumah Sakit, Peraturan Menteri Kesehatan Republik Indonesia Nomor 72 Tahun 2016. Jakarta, Indonesia: Kementerian Kesehatan Republik Indonesia; 2016.
2. Hazarika I. Therapeutic drug monitoring (TDM): an aspect of clinical pharmacology and pharmacy practice. Res Rev A J Pharmacol. 2015;5(3):27–34.
3. Krause KM, Serio AW, Kane TR, Connolly LE. Aminoglycosides: an overview. Cold Spring Harb Perspect Med. 2016;6(6):a027029. CrossRef
4. Pajot O, Burdet C, Couffignal C, Massias L, Armand-Lefevre L, Foucrier A, et al. Impact of imipenem and amikacin pharmacokinetic/pharmacodynamic parameters on microbiological outcome of Gram-negative bacilli ventilator-associated pneumonia. J Antimicrob Chemother. 2015;70(5):1487–94. CrossRef
5. Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of America and the American thoracic society. Clin Infect Dis. 2016;63(5):e61–111. CrossRef
6. Aliska G, Setiabudy R, Purwantyastuti, Karuniawati A, Sedono R, Dewi TU, et al. Optimal amikacin levels for patients with sepsis in intensive care unit of Cipto Mangunkusumo Hospital, Jakarta, Indonesia. Acta Med Indones Indones J Intern Med. 2017;49(3):227–35.
7. Utami ED, Puspitasari I, Asdie RH, Lukitaningsih E, Dahesihdewi A. Amikasin: profil Penggunaan pada Pasien Dewasa Rawat Inap di RSUP Dr. Sardjito Yogyakarta berdasarkan Fungsi Ginjal. J Farm Indones. 2021;13(1):1–12. CrossRef
8. Dana WJ, Fuller MA, Goldman MP, Golembiewsky JA, Gonzales JP, Lowe JF, et al. Drug information handbook. 22nd ed. Hudson, OH: Lexicomp; 2013.
9. American Society of Health-System Pharmacists. American hospital formulary service drug information. 44th ed. Bethesda, MD: American Society of Health-System Pharmacists; 2002.
10. Adaway JE, Keevil BG. Therapeutic drug monitoring and LC-MS/MS. J Chromatogr B. 2012;883–884:33–49. CrossRef
11. Wahyono D. Farmakokinetika Klinik: Konsep Dasar Dan Terapan Dalam Farmasi Klinik. Yogyakarta, Indonesia: Gadjah Mada University Press; 2013.
12. Barco S, Castagnola E, Gennai I, Barbagallo L, Loy A, Tripodi G, et al. Ultra high performance liquid chromatography-tandem mass spectrometry vs. commercial immunoassay for determination of vancomycin plasma concentration in children. Possible implications for everyday clinical practice. J Chemother. 2016;28(5):395–402. CrossRef
13. Lu CY, Feng CH. Micro-scale analysis of aminoglycoside antibiotics in human plasma by capillary liquid chromatography and nanospray tandem mass spectrometry with column switching. J Chromatogr A. 2007;1156(1–2):249–53. CrossRef
14. da Silva ACC, Lizot LDLF, Bastiani MF, Antunes MV, Brucker N, Linden R. Ready for TDM: simultaneous quantification of AMK, vancomycin, and creatinine in human plasma employing ultra-performance liquid chromatography-tandem mass spectrometry. Clin Biochem. 2019;70:39–45. CrossRef
15. Barco S, Mesini A, Barbalgallo L, Maffia A, Tripodi G, Pea F, et al. A liquid chromatography-tandem mass spectrometry platform for routine therapeutic drug monitoring of 14 antibiotics: application to critically ill pediatric patients. J Pharm Biomed Anal. 2020;186:1–7. CrossRef
16. Bijleveld Y, de Haan T, Toersche J, Jorjani S, van der Lee J, Groenendaal F, et al. A simple quantitative method analyzing amikacin, gentamicin, and vancomycin levels in human newborn plasma using ion-pair liquid chromatography/tandem mass spectrometry and its applicability to a clinical study. J Chromatogr B. 2014;951–952:110–8. CrossRef
17. Harmita AAK, Harahap Y, Supandi. Liquid chromatography-tandem mass spectrometry (LC-MS/MS). Jakarta, Indonesia: ISFI Penerbitan; 2019.
18. Chan K, Wang W, Ledesma KR, Yin T, Tam VH. A robust LC-MS/MS method for amikacin: application to cellular uptake and pharmacokinetic studies. Bioanalysis. 2020;12(7):445–54. CrossRef
19. Arsand JB, Jank L, Martins MT, Hoff RB, Barreto F, Pizzolato TM, et al. Determination of aminoglycoside residues in milk and muscle based on a simple and fast extraction procedure followed by liquid chromatography coupled to tandem mass spectrometry and time of flight mass spectrometry. Talanta. 2016;154:38–45. CrossRef
20. Kaufmann A, Butcher P, Maden K. Determination of aminoglycoside residues by liquid chromatography and tandem mass spectrometry in a variety of matrice. Anal Chim Acta. 2012;711:46–53. CrossRef
21. McGlinchey TA, Rafter PA, Regan F, McMahon GP. A review of analytical methods for the determination of aminoglycoside and macrolide residues in food matrice. Anal Chim Acta. 2008;642:1–15. CrossRef
22. Zhu WX, Yang JZ, Wei W, Liu YF, Zhang SS. Simultaneous determination of 13 aminoglycoside residues in foods of animal origin by liquid chromatography-electrospray ionization tandem mass spectrometry with two consecutive solid-phase extraction steps. J Chromatogr A. 2008;1207:29–37. CrossRef
23. Oertel R, Neumeister V, Kirch W. Hydrophilic interaction chromatography combined with tandem-mass spectrometry to determine six aminoglycosides in serum. J Chromatogr A. 2004;1058:197–201. CrossRef
24. European Medicine Agency. Guideline on bioanalytical method validation. London, UK: European Medicine Agency; 2011.
25. Badan Pengawasan Obat dan Makanan. Pedoman Metodologi Uji Bioekivalensi Spesifik Zat Aktif. Jakarta, Indonesia: Badan Pengawasan Obat dan Makanan; 2011.
26. Kahsay G, Song H, Van Schepdael A, Cabooter D, Adams E. Hydrophilic interaction chromatography (HILIC) in the analysis of antibiotics. J Pharm Biomed Anal. 2014;87:142–54. CrossRef
27. Farouk F, Azzazy HME, Niessen WMA. Challenges in the determination of aminoglycoside antibiotics, a review. Anal Chim Acta. 2015;890:21–43. CrossRef
28. Djikstra JA, Sturkenboom MGG, van Hateren K, Koster RA, Greijdanus B, Alffenaar JC. Quantification of amikacin and kanamycin in serum using a simple and validated LC-MS/MS method. Bioanalysis. 2014;6(16):2125–33. CrossRef
29. Chan CC, Lam H, Lee YC, Zang XM. Analytical method validation and instrument performance verification. Hoboken, NJ: John Wiley & Sons; 2004. CrossRef
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website