
INTRODUCTION
Polycystic ovary syndrome (PCOS) is a common gynecological endocrine disorder affecting reproductive-age women. It was first reported in 1935 and was called Stein Leventhal syndrome. In 2003, a diagnostic criterion named Rotterdam criteria was proposed. This includes atleast two of the following three symptoms—hyperandrogenism, oligo-ovulation, and polycystic ovaries to be manifested in PCOS women. The National Institute of Health and Androgen Excess PCOS Society has included an additional diagnosticfactor—hyperinsulinemia, which shows a link to metabolic dysfunction in PCOS [1].
Although the etiopathogenesis of PCOS is not yet clear, one of the main reasons deciphered is hormonal imbalance. High levels of androgens have an effect on the hypothalamus-pituitary axis that leads to changes in the production of luteinizing hormone (LH) and estrogen. These changes cause the symptoms of PCOS, such as anovulation and hyperandrogenism, and might lead to infertility [2]. In most PCOS women, the LH: follicle stimulating hormone ratio is imbalanced, causing high proliferation of theca cells that leads to increased steroidogenesis [3].
During steroidogenesis, aromatase (the rate-limiting enzyme) converts androgens to estrogens [4], while 17 beta-hydroxysteroid dehydrogenase type 1 (17β-HSD1) converts androstenedione to testosterone [5]. Many PCOS women suffer from hyperandrogenism because of excess production of androgens and altered androgen receptor (AR) signaling pathways [6]. Some PCOS women manifest high estrogen levels, and their actions will be mediated by estrogen alpha and beta receptors [7]. Currently, letrozole, abiraterone, flutamide, and tamoxifen are used as effective antagonists for aromatase, 17β-HSD1, androgen, and estrogen receptors for reducing the estrogens and androgens in PCOS women [8–10].
Considering the side effects of allopathic drugs, it is important to identify an alternative treatment [11]. In recent years, green medicine or herbal medicine implicated in PCOS treatment showed less toxic side effects and are effective due to the presence of various bioactive compounds. In a recent study, the use of Apium graveolens supplements and Eucalyptus globulus essential oil showed a significant reduction of stress in reproductive women and improved their folliculogenesis [12]. These bioactive compounds, useful in the creation of novel medications, have the advantage of being low-cost and high-efficiency [13].
Saraca asoca (Roxb.) Willd., commonly known as Ashoka, is the most widely found ancient medicinal plant in India belonging to the Family Caesalpiniaceae [14]. In India, Ashoka is one of the typical medicinal plants for treating gynecological diseases because of its stimulating effect on the endometrium [15]. Saraca asoca has many pharmacological properties such as anti-cancer, anti-bacterial, anti-diabetic, hypolipidemic, anti-inflammatory, estrogenic, and anti-mennorhagic [14]. As Ashoka is used for many gynecological issues, ayurvedic practitioners recommend this plant for PCOS. Ashokarishta, one of the ayurvedic formulations used for PCOS treatment, contains bark as the main ingredient [16].
In this study, in-silico tools were used for screening potential anti-androgen and anti-estrogenic compounds from the bark and flowers of S. asoca. For this, 56 compounds previously reported from this plant were docked against targets, such as aromatase, 17β-HSD1, androgen, and estrogen receptors (α and β), to find the potential bioactive compounds for use against PCOS.
MATERIALS AND METHODS
Selection of proteins
Five targets (aromatase, 17β hydroxysteroid dehydrogenase type 1, androgen, and estrogen α and estrogen β receptors) that have a direct effect on androgen and estrogen biosynthesis and signaling were chosen from the ovarian steroidogenesis pathway.
Preparation of proteins
3-D crystallographic protein structures for the respective proteins of human origin were retrieved from the protein data bank (PDB) (https://www.rcsb.org/) and downloaded in the PDB format. Five proteins chosen were (1) human aromatase (CYP19A1) (PDB ID: 3S79), (2) human 17β- hydroxysteroid dehydrogenase type 1 (17β-HSD1) (PDB ID: 1FDS), (3) human AR (PDB ID: 1E3G), (4) human estrogen α receptor (PDB ID: 3ERT), and (5) human estrogen β receptor (PDB ID: 2QTU).
PyMol is an open-source molecular visualization software used to 3-D structures of macromolecules and to check the interactions of hydrogen bonds between the target and ligand [17]. With the help of Auto Dock, the water molecules and other ligands present in each target structure were removed, and additional charges, such as Kollman charges, were added to the proteins. Then, the structures were downloaded in pdbqt format for use in future docking operations [18,19].
Ligand selection
Research articles published in PubMed and SCOPUS were screened for the bioactive compounds (ligands) from S. asoca. Previously documented bioactive compounds from various parts of S. asoca identified by different chromatographic techniques were chosen and screened for molecular docking. Fifty-six compounds found in the bark and flowers of S. asoca were chosen for further investigation. For each target, the top commercially available inhibitors used were obtained and chosen for docking. A comparison of bioactive compounds and commercial inhibitors was checked to see the interaction of protein and ligands in terms of binding score, hydrogen, and hydrophobic interactions.
Preparation of ligands
PubChem database [https://pubchem.ncbi.nlm.nih.gov/] was used to retrieve the 3-D structures of the selected compounds. Open Babel is a free chemical toolbox that converts the spatial data file format files to PDB format files [20]. The angles, charges, force field, and torsion roots for each ligand were determined and prepared based on these parameters. The ligand structures were finally converted to pdbqt format for the docking process.
Active site prediction
Prior to docking, a suitable active site for each protein must be identified since ligands will bind near active sites, and they are identified using the coordinates of native ligands present in the target protein that is retrieved from the PDB. The active site residues were discovered to be atoms at less than 25 A°, admitting the ligand to bind in that position [21]. Supercomputing Facility for Bioinformatics and Computational Biology at the Indian Institute of Technology, Delhi (http://www.scfbio-iitd.res.in/dock/activesite.jsp), was used to find the active sites of the chosen targets.
Molecular docking
Molecular docking was performed by PyRx’s AutoDock Vina. To select the targeted protein and ligands, Vina wizard control was used, and a grid was displayed on the selected target [22], and to consider the active sites, the grid was adjusted to get better docking sites. Autodock Vina performed the docking after the grid was selected. As a result, the binding affinity of each ligand was determined.
Density functional theory (DFT)
DFT is a computational quantum mechanical modeling tool that is mainly used to check the chemical activity and correlate the calculated energies. Gaussian 09 6-31G (d, p) basis set and B3LYP method were used to calculate the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), total energy, energy gap, and chemical potential. Global descriptors, including electrophilicity index, electronegativity, absolute hardness, and softness, describe the chemical behavior of the molecules were calculated [23].
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties
Drug discovery with novel targets and effective biologically active compounds is aided by ADMET properties. In addition, physiochemical and pharmacokinetic parameters are also predicted [24]. ADMET properties of the chosen ligands were predicted using the ADMET Lab server 2.0 tool (https://admetmesh.scbdd.com) This program performs numerous drug-likeness evaluations and finds ADMET-related features [25].
RESULTS AND DISCUSSION
Selection of ligands
Fifty-six bioactive compounds from the bark and flower parts of S. asoca reported in the literature were selected for molecular docking [26–35] (Supplementary Table 1). Commercial ligands, such as letrozole, abiraterone, flutamide, and tamoxifen, were docked against aromatase, 17β-HSD1, androgen, and estrogen receptors, respectively.
Active site prediction
SCFBio tool predicted the active sites of human aromatase (PDB ID: 3S79), human 17β-hydroxysteroid dehydrogenase type 1 (PDB ID: 1FDS), human AR (PDB ID: 1E3G), human estrogen α receptor (PDB ID: 3ERT), and human estrogen β receptor (PDB ID: 2QTU). Table 1 shows the amino acid residues present at the predicted active sites.
Molecular docking
All five receptors were docked against 56 selected compounds reported from S. asoca bark and flower to check the inhibitory activity. Docking parameters are value-root-mean-square deviation value, ligand and protein complex, hydrogen and hydrophobic interaction, and mainly binding energy of each compound docked against individual proteins. From the docking results, the top three ligands for each protein were chosen and further analyzed. The binding energy of all the fifty-six docked compounds against each protein is given in Supplementary Table 2. The results of docking calculation in terms of binding affinity (kcal/mol) and hydrogen and hydrophobic interactions of the top three compounds are shown in Table 2.
Aromatase (PDB ID-3S79) bound procyanidin B2 (−11.7 kcal/mol), procyanidin B1 (−11 kcal/mol), and β-sitosterol (−9.8 kcal/mol) with high binding affinity. All these compounds showed hydrogen bond interaction with amino acid residues—ILE-133, THR-310, and SER- 314. The docking poses of the top three ligands are shown in Figure 1; each ligand is represented in different colors to get better visualization. Letrozole, one of the most commonly used aromatase inhibitors, showed −8.2 kcal/mol binding energy against aromatase. TRP-141, THR-310, CYS-437, and ALA-438 exhibited hydrogen bond interaction between letrozole and aromatase. This was also observed in another study, wherein, aromatase (PDB ID-3EQM) docked against letrozole showed −8.7 kcal/mol binding affinity [36]. Thus, letrozole has less binding affinity when compared to the bioactive compounds procyanidin B2, procyanidin B1 and β-sitosterol. β-sitosterol, a phytoestrogen present in flowers of S. asoca, has the potential to regulate estrogen synthesis, implying its role in aromatase [34]. These results show that the bioactive compounds have a good binding affinity and, hence, might have more significant aromatase inhibitory activity than letrozole. A balance in the aromatase activity will induce ovulation in PCOS women and also result in normal androgen levels [4].
17β-HSD1 protein (PDB ID-1FDS) showed highest binding energy of −10.4, −10.1, and −9.2 kcal/mol, respectively, with procyanidin B2, amyrin, and procyanidin B1 interacting at ILE-14, LEU-95, CYS-185, ASN-152, THR-190, TYR-218, and SER-222 amino acids residues. The docking poses of the top three ligands when docked with 17β-HSD1 are shown in Figure 2. Abiraterone, a commercial inhibitor for HSD, when docked against 17β-HSD1, showed a binding score of −9.3 kcal/mol and the interaction of hydrogen bonds with amino acid residue-HIS-221. 17β-HSD1 facilitates the reduction of estrone to estradiol [37]. Thus, the inhibitory action of 17β-HSD1 has an effect on estradiol production. From the results, it is evident that procyanidins and amyrin from S. asoca have the highest binding affinity to 17β-HSD1compared to that of the commercial ligand.
After docking, the AR, (PDB ID- 1E3G) showed good binding energy with leucopelargonidin (−10 kcal/mol), leucocyanidin (−9.8 kcla/mol), and kaempferol (−9.6 kca/mol). ASN-705, GLN-711, MET-745, and ARG-752 amino acids showed hydrogen bond interactions with all the top three binding energy compounds. The hydrogen bond interactions of the top three ligands are represented in Figure 3. Flutamide, a commercial inhibitor of AR, bound with a binding score of −8.1 kcal/mol with the amino acid residues—MET-745 and ARG-752. In other studies, flutamide presented a score of −8.69 kcal/mol against the AR [38]. Androgen excess causes hyperandrogenism, hirsutism, acne, and androgenic alopecia in PCOS women [39]. Thus, to reduce these symptoms, androgen-inhibitory drugs with fewer side effects should be used. Hence, anthocyanidins, such as leucopelargonidin and leucocyanidin, might be potential drugs for blocking ARs and hence further downstream signaling.
 | Table 1. Active sites of each target protein are selected from the ovarian steroidogenesis pathway. [Click here to view] |
 | Table 2. Docking score, hydrogen and hydrophobic interactions of top three ligands. [Click here to view] |
Amongst the 56 compounds, epicatechingallate (−8.9 kcal/mol), ellagic acid (−8.4 kcal/mol), and catechin (−8.4 kcal/mol) revealed good binding affinity against estrogen alpha receptor (PDB ID-3ERT). GLU-419 and HIS-524 amino acids showed hydrogen bond interactions with the estrogen alpha receptor. The docking poses of the top three ligands when docked with estrogen alpha receptor are shown in Figure 4. Estrogen beta receptor (PDB ID-2QTU) had greater binding affinities with leucopelargonidin (−9.1 kcal/mol), leucocyanidin (−9 kcal/mol), and luteolin (−9 kcal/mol). GLU-305, ARG-346, and GLY-472 amino acids showed greater hydrogen bonding with the bioactive compounds. The hydrogen and hydrophobic interactions of the top three ligands of estrogen beta receptor are displayed in Figure 5. Tamoxifen, a commercial ligand for estrogen, revealed a binding score of −9.7 kcal/mol. Leucopelargonidin, leucocyanidin, and luteolin compounds showed a closer binding affinity with the commercial ligands after docking with estrogen receptors. PCOS women have estrogen dominance that occurs because of the abnormal function of estrogen and estrogen receptors [40]. Thus, the natural compounds might be helpful in addressing estrogen dominance with fewer side effects.
From these docking results, seven compounds (procyanidin B2, leucopelargonidin, epicatechingallate, amyrin, procyanidin B1, leucocyanidin, and ellagic acid) with high binding energy were selected for further analysis.
DFT calculations
DFT is a computational tool for evaluating the compounds’ nature and molecular structures by calculating their electron density [41]. Chemical reactivity parameters were calculated by using the frontier orbitals such as HOMO and LUMO [42]. Band gap represents the energy and has a direct relation with molecular reactivity (Egap = ELUMO- EHOMO), and all these calculations were determined using B3LYP/6–31G (d, p) basis set. Parameters, such as chemical hardness, electronegativity, electrophilicity index, electronic energy, and chemical potential of the compounds, were calculated for the selected compounds [43]. The statistics of DFTtheory-based molecular descriptors for the selected seven compounds are given in Table 3.
 | Figure 1. (a) The binding poses of procyanidin B2 (blue), procyanidin B1 (pink), and beta-sitosterol (magenta) when docked with aromatase. (b) Procyanidin B2 interacting residues, ILE-133, TRP-141, THR-310, SER-314, and ARG-435 (green) form hydrogen bonds (yellow) of length 2.0 Å, 1.8 Å, 1.8 Å, 2.4 Å, and 2.3Å, respectively. (c) Procyanidin B1 interacting residues, ILE-133, ARG-145, TRP-141, THR-310, SER-314, and ARG-435 (green) form hydrogen bonds (yellow) of length 2.0 Å, 2.3 Å, 1.8 Å, 1.8 Å, 2.5 Å and 2.3 Å, respectively. (d) β-sitosterol did not show any hydrogen interactions. [Click here to view] |
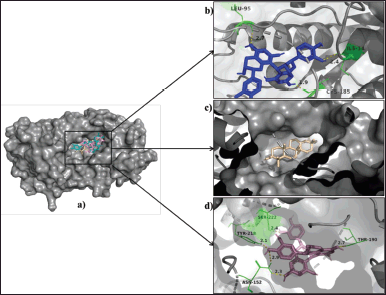 | Figure 2. (a) Binding poses of procyanidin B2 (blue), amyrin (wheat), and procyanidin B1 (pink) in the 17β-HSD1. (b) Procyanidin B2 interacting residues, ILE-14, LEU-95, and CYS-185 (green), form hydrogen bonds (yellow) of length 2.4 Å, 2.7 Å, and 1.9 Å, respectively. (c) Amyrin interacting residues did not show any hydrogen interactions. (d) Procyanidin B1 interacting residues, ASN-152, THR-190, TYR-218, and SER-222 (green) form hydrogen bonds (yellow) of length 2.3 Å, 2.9 Å, 2.7 Å, 2.1 Å and 2.4 Å, respectively. [Click here to view] |
Ellagic acid (0.15864eV), epicatechingallate (0.16934 eV), and procyanidin B2 (0.19387 eV) compounds have less energy gap, which shows that these compounds are soft molecules. Procyanidin B2, with a value of −0.19527 eV, exhibits the highest HOMO showing this compound has the best electron donor. Ellagic acid (−0.0691 eV) has the lowest LUMO, showing that this compound has the best electron acceptor. Electronegativity determines the chemical behavior of a compound [44]. In our study, ellagic acid showed greater electronegativity value. The electronegativity also talks about the inhibition effect of a molecule [45].
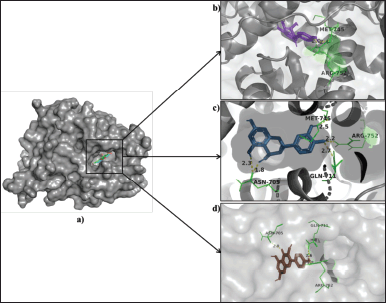 | Figure 3. (a) Binding poses of leucopelargonidin (purple), leucocyanidin (skyblue), and kaempferol (brown) in the AR. (b) Leucopelargonidin interacting residues, MET-745, ARG-752 (green), form hydrogen bonds (yellow) of length 2.4 Å and 2.0 Å, respectively. (c) Leucocyanidin interacting residues, ASN-705, GLN-711, MET-745 (green) form hydrogen bonds (yellow) of length 1.8 Å, 2.3 Å, 2.7 Å, and 2.5 Å, respectively. (d) Kaempferol interacting residues, ASN-705, GLN-711, and ARG-752 (green), form hydrogen bonds (yellow) of length 2.9 Å, 2.6 Å, and 1.8 Å, respectively. [Click here to view] |
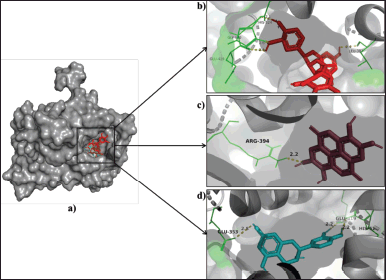 | Figure 4. (a) Binding poses of epicatechingallate (red), ellagic acid (brown), and catechin (cyan) in the estrogen alpha receptor. (b) Epicatechingallate interacting residues, HIS-524, GLU-419, GLY-420, LEU-387 (green), form hydrogen bonds (yellow) of length 2.6 Å, 2.1 Å, 2.4 Å, and 2.4 Å, respectively. (c) Ellagic acid interacting residues, ARG-394 (green), form hydrogen bonds (yellow) of length 2.2 Å. (d) Catechin interacting residues, GLU-419, GLU-353, HIS-524 (green), form hydrogen bonds (yellow) of length 2.2 Å, 2.8 Å, and 2.2 Å, respectively. [Click here to view] |
Procyanidin B1 (7.914611) has the greater dipole moment, followed by procyanidin B2 and leucopelargonidin. The dipole moment is directly proportional to chemical reactivity. The chemical stability and reactivity of a compound were determined by chemical hardness [42]. Procyanidin B2 and procyanidin B1 have a lower chemical hardness which shows these compounds have good stability. Chemical reactivity, stability, nature, and optimized structures of compounds were determined using a DFT study, and these results are compatible with the docking results. The optimized and HOMO-LUMO structures of the selected seven ligands are given in Figure 6.
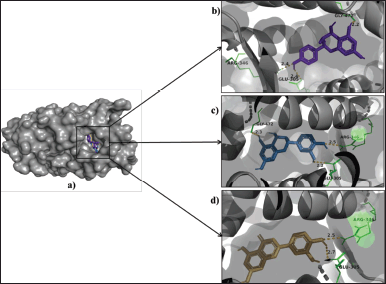 | Figure 5. (a) Binding poses of leucopelargonidin (purple), leucocyanidin (blue), and luteolin (light orange) in the estrogen beta receptor. (b) Leucopelargonidin interacting residues, GLU-305, ARG-346, GLY-472 (green) form hydrogen bonds (yellow) of length 2.4 Å, 2.4 Å, and 2.2 Å, respectively. (c) Leucocyanidin interacting residues, GLY-472, ARG-346 (green), form hydrogen bonds (yellow) of length 2.3 Å and 2.5 Å, respectively. (d) Luteolin interacting residues GLU-305, ARG-346 (green) form hydrogen bonds (yellow) of length 2.7 Å and 2.5 Å, respectively. [Click here to view] |
ADMET prediction
In-silico ADMET analysis is a rapid way to identify if a molecule has adequate pharmacokinetics and pharmacodynamics properties. In the present study, seven bioactive compounds with top docking scores were selected to predict the ADMET properties. ADME, and physiochemical properties of the bioactive compounds are represented in Table 4.
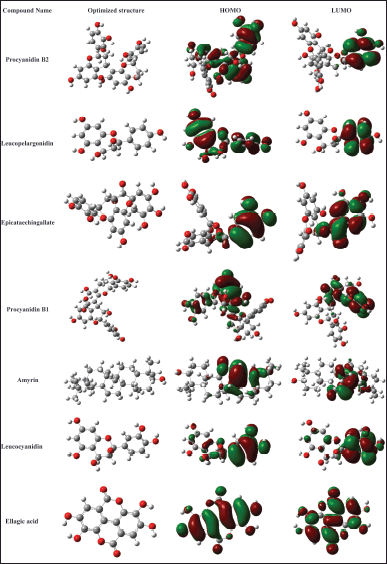 | Figure 6. HOMO-LUMO and optimized structures of the selected seven ligands. [Click here to view] |
Since cytochrome P450 (CYP) is involved in drug metabolism, this parameter was investigated [46]. In this study, all the compounds exhibited as substrates with better binding results, thus showing that these compounds will be easier for the metabolism. All seven compounds exhibited less than 3 hours of half-life in excretion properties. Of all the compounds, procyanidin B2 showed significantly less half-life while ellagic acid showed a higher half-life, indicating that these compounds will easily be eliminated from the body, and all the seven compounds showed as nonblockers of AMES and hERG.
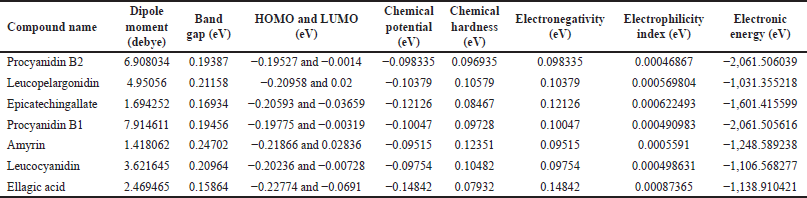 | Table 3. DFT calculations of seven selected ligands. [Click here to view] |
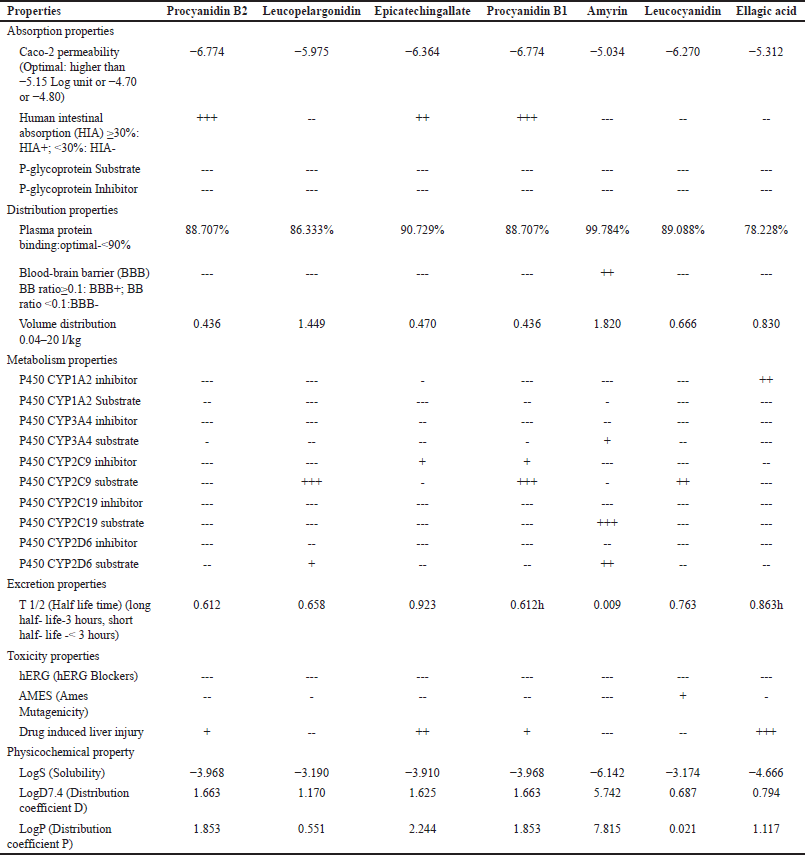 | Table 4. ADMET properties of the selected ligands. [Click here to view] |
Before drug design, the compounds should be checked for the pharmacokinetics and toxicity. In addition to being effective against the therapeutic target, a high-quality drug candidate should also pass the required ADMET properties at a therapeutic dose. Thus, ADMET properties play a key role in the drug development process [47].
CONCLUSION
PCOS is a metabolic and endocrine disorder that affects 6%–15% of women of reproductive age. Because herbal medicine has fewer side effects, medicinal plants have been used to treat PCOS symptoms. Saraca asoca, which contains a diverse range of phytochemicals, is a commonly used plant for treating PCOS symptoms in India. Of all the selected compounds, procyanidin B2 and leucopelargonidin showed the highest binding affinity with proteins, such as aromatase, 17β-HSD1, androgen, and estrogen receptors of ovarian steroidogenesis pathway. In addition, these compounds demonstrated promising DFT and ADMET properties. Thus, we propose procyanidin B2 and leucopelargonidin as promising compounds in the management of PCOS with hyperandrogenism and estrogen dominance. Further in-vivo and in-vitro studies are needed to determine the inhibitory and toxic effects of these compounds for the discovery of drugs against many gynecological disorders.
ACKNOWLEDGMENT
The authors wish to thank Dr. Thirumurthy M, for his support in the docking analysis. The authors are grateful to the facility given by SRM Institute of Science and Technology to carry out the work.
LIST OF ABBREVIATIONS
17β-HSD1, 17 beta-hydroxysteroid dehydrogenase type 1; ADMET, Absorption, distribution, metabolism, excretion, and toxicity; CYP19A1, Human aromatase; DFT, Density functional theory; HOMO, Highest occupied molecular orbital; LH, Luteinizing hormone; LUMO, Lowest unoccupied molecular orbital; PCOS, Polycystic ovary syndrome; PDB, Protein data bank.
AUTHORS’ CONTRIBUTIONS
HK, VK, and UB originated and outlined experiments; HK and VK conducted the experiments and interpreted the data. HK and VK original draft preparation; UB conceptualization, supervision, writing, review, and editing. All the authors contributed to the manuscript preparation.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest.
ETHICAL APPROVAL
This study does not involve the use of animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7(4):219–31. doi: https://doi.org/10.1038/ nrendo.2010.217
2. Oyebanji OG, Asaolu MF, Amonimo EO. Hormonal imbalance in polycystic ovarian syndrome (PCOS) in Teaching Hospitals in Ekiti State, Nigeria. Open J Obstet Gynecol. 2018;08(13):1456–64. doi: https://doi.org/10.4236/ojog.2018.813147
3. Ashraf S, Nabi M, Rasool SA, Rashid F, Amin S. Hyperandrogenism in polycystic ovarian syndrome and role of CYP gene variants: a review. Egypt J Med Hum Genet. 2019;20:25. doi: https://doi. org/10.1186/s43042-019-0031-4
4. Chen J, Shen S, Tan Y, Xia D, Xia Y, Cao Y, et al. The correlation of aromatase activity and obesity in women with or without polycystic ovary syndrome. J Ovarian Res. 2015;8(1):4–9. doi: https://doi. org/10.1186/s13048-015-0139-1
5. Aka JA, Zerradi M, Houle F, Huot J, Lin SX. 17Beta-hydroxysteroid dehydrogenase type 1 modulates breast cancer protein profile and impacts cell migration. Breast Cancer Res. 2012;14(3):R92. doi: https:// doi.org/10.1186/bcr3207
6. Gao XY, Liu Y, Lv Y, Huang T, Lu G, Liu HB, et al. Role of androgen receptor for reconsidering the “True” polycystic ovarian morphology in PCOS. Sci Rep. 2020;10(1):1–7. doi: https://doi.org/10.1038/s41598- 020-65890-5
7. Walters KA. Polycystic ovary syndrome: is it androgen or estrogen receptor? Curr Opin Endocr Metab Res. 2020;12:1–7. doi: https:// doi.org/10.1016/j.coemr.2020.01.003
8. Guang HJ, Li F, Shi J. Letrozole for patients with polycystic ovary syndrome: a retrospective study. Medicine. 2018;97(44):e13038. doi: https://doi.org/10.1097/MD.0000000000013038
9. De LV, Lanzetta D, Antona D, La MA, Morgante G. Hormonal effects of flutamide in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:99–102. doi: https://doi.org/10.1210/ jcem.83.1.4500
10. Dhaliwal KL, Suri V, Gupta KR, Sahdev S. Tamoxifen: an alternative to clomiphene in women with polycystic ovary syndrome. J Hum Reprod Sci. 2011;4:76–9. doi: https://doi.org/10.4103/0974-1208.86085
11. Abasian Z, Rostamzadeh A, Mohammadi M, Hosseini M, Rafieian-kopaei M. A review on role of medicinal plants in polycystic ovarian syndrome: pathophysiology, neuroendocrine signaling, therapeutic status and future prospects. Middle East Fertil Soc J. 2018; 23:255–62. doi: https://doi.org/10.1016/j.mefs.2018.04.005
12. Novika RGH, Nurhidayati S, Wahidah NJ, Maulina R, Sumarno L, Yunus A, et al. The effects of Apium graveolens and Eucalyptus globulus in decreasing stress and protecting folliculogenesis marker on woman reproductive health during COVID-19 pandemic. Indones J Pharm. 2022;33:592–601. doi: https://doi.org/10.22146/ijp.4511
13. Prokopenko YS, Perekhoda LO, Georgiyants VA. Docking stuides of biologically active substances from plant extracts with anticonvulsant activity. J Appl Pharm Sci. 2019;9:66–72. doi: http://doi.org/10.7324/ JAPS.2019.90110
14. Singh S, Anantha Krishna TH, Kamalraj S, Kuriakose GC, Valayil JM, Jayabaskaran C. Phytomedicinal importance of Saraca asoca (Ashoka): an exciting past, an emerging present and a promising future. Curr Sci. 2015;109(10):1790–801. doi: https://doi.org/10.18520/v109/i10/1790-1801
15. Bhalerao SA, Verma DR, Didwana VS, Teli NC. Saraca asoca (Roxb.), De. Wild: an overview. Ann Plant Sci. 2014;3(7):770–5.
16. Mishra S, Meshram PS, Tawalare K. Critical appraisal on PCOD/PCOS and its treatment in ayurveda and allopathy. Int J Pharm Biol Sci. 2018;9:130–5.
17. Yuan S, Chan HCS, Hu Z. Using PyMOL as a platform for computational drug design. WIREs Comput Mol Sci. 2017;7:1–10. doi: https://doi.org/10.1002/wcms.1298
18. Angadi KK, Gundampati RK, Jagannadham MV, Kandru A. Molecular docking studies of guggultetrol from Nymphaea pubescens with target glucokinase (GK) related to type-ii diabetes. J Appl Pharm Sci. 2013;3:127–31. doi: https://doi.org/10.7324/JAPS.2013.30222
19. Gundampati RK, Jagannadham MV. Molecular docking based inhibition of trypanothione reductase activity by Taxifolin novel target for antileishmanial activity. J Appl Pharm Sci. 2012;2:133–6. doi: https://doi.org/10.7324/JAPS.2012.21026
20. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminformatics. 2011;3:33. doi: https://doi.org/10.1186/1758-2946-3-33
21. Ashraf Z, Saeed A, Nadeem H. Design, synthesis and docking studies of some novel isocoumarin analogues as antimicrobial agents. RSC Adv. 2014;4:53842–53. doi: https://doi.org/10.1039/c4ra07223e
22. Alamri MA, Alamri MA. Pharmacophore and docking-based sequential virtual screening for the identification of novel sigma 1 receptor ligands. Bioinformation. 2019;15:586–95. doi: https://doi. org/10.6026/97320630015579
23. Deghady AM, Hussein RK, Alhamzani AG, Mera A. Article density functional theory and molecular docking investigations of the chemical and antibacterial activities for 1-(4-hydroxyphenyl)-3-phenylprop-2-en-1-one. Molecules. 2021;26(12):3631. doi: https://doi.org/10.3390/molecules26123631
24. Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–105. doi: https://doi.org/10.1021/ci300367a
25. Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–14. doi: https://doi.org/10.1093/nar/gkab255
26. Tewari R, Gupta M, Ahmad F, Rout PK, Misra L, Patwardhan A, et al. Extraction, quantification and antioxidant activities of flavonoids, polyphenols and pinitol from wild and cultivated Saraca asoca bark using RP-HPLC-PDA-RI method. Ind Crops Prod. 2017;103:73–80. doi: http://dx.doi.org/10.1016/j.indcrop.2017.03.036
27. Sadhu SK, Khatun A, Phattanawasin P, Ohtsuki T, Ishibashi M. Lignan glycosides and flavonoids from Saraca asoca with antioxidant activity. J Nat Med. 2007;61(4):480–2.
28. Gahlaut A, Shirolkar A, Hooda V, Dabur R. A rapid and simple approach to discriminate various extracts of Saraca asoca [Roxb.], De. Wild using UPLC-QTOFMS and multivariate analysis. J Pharm Res. 2013;7(2):143–9. doi: http://dx.doi.org/10.1016/j.jopr.2013.03.005
29. Saha J, Mukherjee S, Gupta K, Gupta B. High-performance thin-layer chromatographic analysis of antioxidants present in different parts of Saraca asoca (Roxb.) de Wilde. J Pharm Res. 2013;7(9):798–803. doi: http://dx.doi.org/10.1016/j.jopr.2013.10.004
30. Shirolkar A, Gahlaut A, Chhillar AK, Dabur R. Quantitative analysis of catechins in Saraca asoca and correlation with antimicrobial activity. J Pharm Anal. 2013;3(6):421–8. doi: http://dx.doi.org/10.1016/j.jpha.2013.01.007
31. Saravanan S, Babu NP, Pandikumar P, Ignacimuthu S. Therapeutic effect of Saraca asoca (Roxb.) Wilde on lysosomal enzymes and collagen metabolism in adjuvant induced arthritis. Inflammopharmacology. 2011;19(6):317–25.
32. Ahmad F, Misra L, Tewari R, Gupta P, Mishra P, Shukla R. Anti-inflammatory flavanol glycosides from Saraca asoca bark. Nat Prod Res. 2016;30(4):489–92.
33. Joshi RK. E, e - α -Farnesene rich essential oil of Saraca asoca (Roxb.) Wilde flower. Nat Prod Res. 2016;30(8):979–81.
34. Swar G, Shailajan S, Menon S. Activity based evaluation of a traditional ayurvedic medicinal plant: Saraca asoca (Roxb.) de Wilde flowers as estrogenic agents using ovariectomized rat model. J Ethnopharmacol. 2017;195:324–33. doi: http://dx.doi.org/10.1016/j.jep.2016.11.038
35. Mishra S, Aeri V. Biotransformation of lignan glycoside to its aglycone by Woodfordia fruticosa flowers: quantification of compounds using a validated HPTLC method. Pharm Biol. 2017;55(1):360–6. doi: http://dx.doi.org/10.1080/13880209.2016.1238948
36. Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular docking of aromatase inhibitors. Molecules. 2011;16:3597–617. doi: https://doi.org/10.3390/molecules16053597
37. Hilborn E, Stål O, Jansson A. Estrogen and androgen-converting enzymes 17β-hydroxysteroid dehydrogenase and their involvement in cancer: with a special focus on 17β-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget. 2017;8:30552–62. doi: https://doi.org/10.18632/oncotarget.15547
38. Singh AN, Baruah MM, Sharma N. Structure based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against prostate cancer. Sci Rep. 2017;7:1–8. doi: https://doi.org/10.1038/s41598-017-02023-5
39. Paris VR, Bertoldo MJ. The mechanism of androgen actions in PCOS etiology. Med Sci. 2019;7:89.
40. Xu XL, Deng SL, Lian ZX, Yu K. Estrogen receptors in polycystic ovary syndrome. Cells. 2021;10:1–13. doi: https://doi.org/10.3390/ cells10020459
41. Abbaz T, Bendjeddou A, Villemin D. Molecular orbital studies (hardness, chemical potential, electro negativity and electrophilicity of TTFs conjugated between 1, 3-dithiole. Int J Adv Res Sci Eng Technol. 2018;5:5150–61.
42. AlShamaileh E. DFT study of monochlorinated pyrene compounds. Comput Chem. 2014;02:43–9. doi: https://doi.org/10.4236/ cc.2014.23006
43. Raftani M, Abram T, Azaid A, Kacimi R, Bennani MN, Bouachrine M. Theoretical design of new organic compounds based on diketopyrrolopyrrole and phenyl for organic bulk heterojunction solar cell applications: DFT and TD-DFT study. Mater Today: Proc. 2021;45:7334–43. doi: https://doi.org/10.1016/j.matpr.2020.12.1228
44. Kerru N, Gummidi L, Bhaskaruni SVHS, Maddila SN, Singh P, Jonnalagadda, SB. A comparison between observed and DFT calculations on structure of 5-(4-chlorophenyl)-2-amino-1,3,4- thiadiazole. Sci Rep. 2019;9:1–17. doi: https://doi.org/10.1038/ s41598-019-55793-5 21
45. Yadalam PK, Varatharajan K, Rajapandian K, Chopra P, Arumuganainar D, Nagarathnam T, et al. Antiviral essential oil components against SARS-CoV-2 in pre-procedural mouth rinses for dental settings during COVID-19: a computational study. Front Chem. 2021;9:1–11.
46. Lagorce D, Douguet D, Miteva MA, Villoutreix BO. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci Rep. 2017;7:1–15. doi: https://doi.org/10.1038/srep46277
47. Guan L, Yang H, Cai Y, Sun L, Di P, Li W, et al. ADMET-score-a comprehensive scoring function for evaluation of chemical drug-likeness. Med Chem Comm. 2019;10:148–57. doi: https://doi. org/10.1039/C8MD00472B
SUPPLEMENTARY MATERIALS
 | Supplementary Table 1. Compounds that are selected from the literature for molecular docking. [Click here to view] |
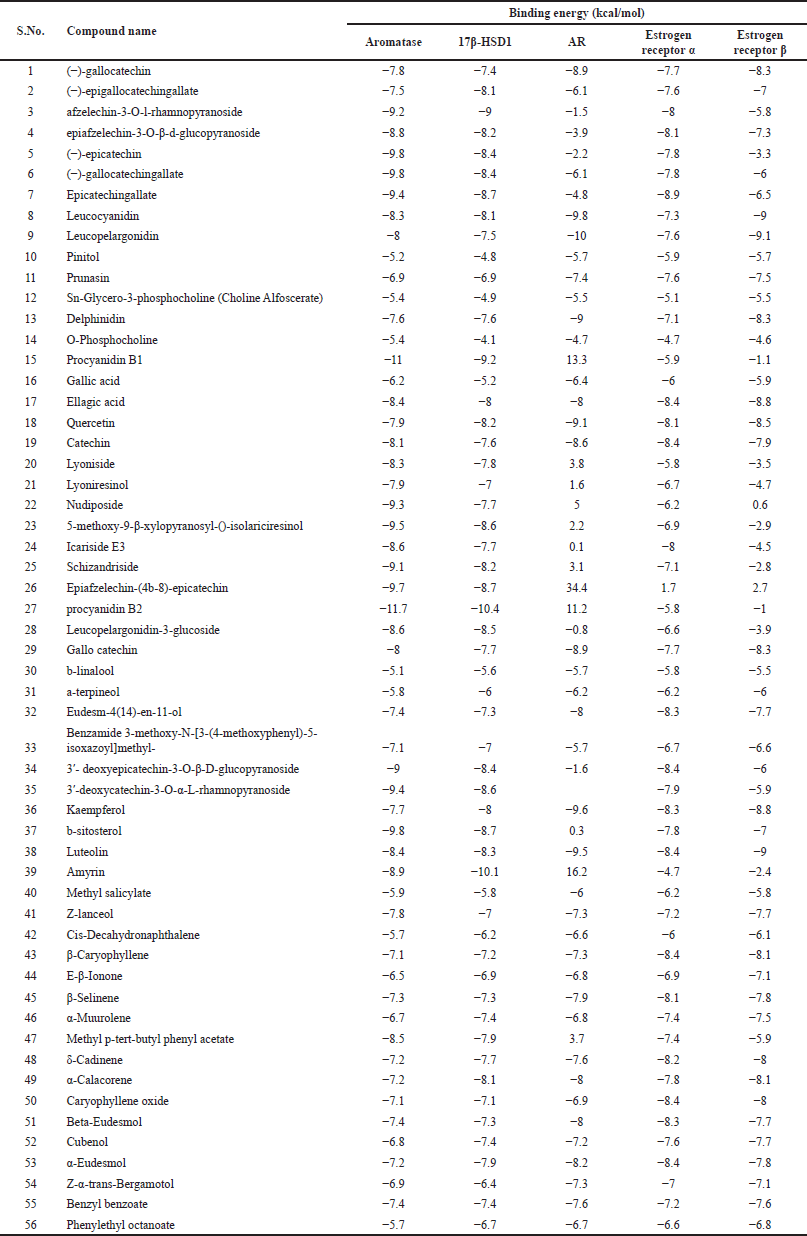 | Supplementary Table 2. Binding energy of all the docked compounds against each target protein. [Click here to view] |