
INTRODUCTION
Breast cancer is a collection of illnesses wherein an individual’s breast tissue’s cells alter and divide out of control, frequently producing a lump or mass. Mammography is essential to prompt detection since breast cancer often has little or no symptoms when the tumor is small and most amenable to treatment. A bulge or lump is the most typical physical symptom. Even before the first breast tumor is large enough to be sensed, breast cancer can occasionally spread to the lymph nodes underneath the arm and generate a bump or inflammation [1].
Men and women are both affected, but women experience it more frequently. Breast cancer, besides being the most frequently identified cancer among American women, is the second-leading cause of cancer-related fatality for women in the US behind lung cancer. About one in eight women will be diagnosed with breast cancer over their lifetime. According to the most recent data, the overall relative survival rates for women with breast cancer are as follows: 91% at 5 years, 84% at 10 years, and 80% at 15 years. There are more than 3.8 million breast cancer survivors including women still being treated and those who have completed treatment [2].
There are often five distinct treatment choices, and most treatment plans incorporate surgeries, radiotherapy, hormonal treatment, chemotherapy, and targeted medications. Some are localized, primarily concentrating on the area around the tumor [3]. Targeted therapy, a category of anticancer therapy, tackles the proteins that determine how cancer cells proliferate, split, and migrate. It forms the cornerstone of precision medicine. As expertise about the DNA variants and proteins that feed cancer improves, researchers are more capable of developing cancer therapies that target these proteins. Several targeted therapies utilize small-molecule drugs or monoclonal antibodies. A few monoclonal antibodies recognize tumor cells, making it easier for the immune system to locate and eradicate them. Certain monoclonal antibodies actively impede the development of cancer cells or cause them to perish [4].
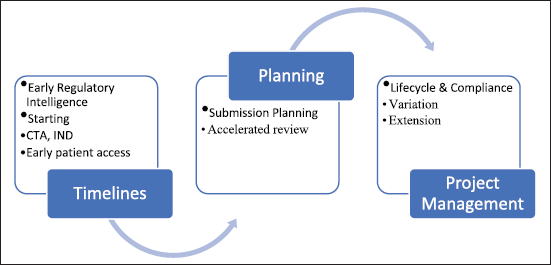 | Figure 1. PLM of a pharmaceutical drug product [8]. [Click here to view] |
The management and outcomes of clinical investigations have long been the focus of drug development. The marketplace is now considering more comprehensive initiatives to improve the procedures for bringing novel drugs onto the market, which can hasten product development while reducing expenses [5]. The term “product lifecycle management” (PLM) describes the cyclical stages of a product, from conception through retirement after development, introduction, and sales. Figure 1 explains Product Lifecycle Management of pharmaceutical drug product from Research through development, authorization and post-approval. This gives an idea on how to plan, divide time per phase and focus on project management. PLM combines the organization’s vision for managing and carrying out broad product planning. PLM decreases development costs and expedites the time to market for new products [6].
At every stage of the product life cycle, a business executes strategies and fluctuates depending on how the market is responding to a product. The product life cycle is significantly influenced by market acceptability, ease of admission into the marketplace, pace of industry advancement, and changes in consumer preferences. When it is easier for rivals to enter markets, consumers frequently shift their opinions about the products they purchase, or the market becomes swiftly saturated, products are more prone to have shortened life cycles during a product life cycle [7].
Regulatory challenges are any roadblocks in the process of clinical trials that lead to delays in their completion and ultimately lead to longer duration to gain approvals, be it for marketing or postmarketing changes. Pipelines are becoming narrower, and the patent term on active revenue-generating drugs is rapidly approaching its end. To reduce the time and expense of clinical development, novel solutions are required to maintain the pharmaceutical industry’s viability [9]. The current article is an attempt to summarize the life cycle stages of blockbuster molecules used in the treatment of breast cancer from a regulatory standpoint, which will help in efficient forecasting and reducing cost.
MATERIALS AND METHODS
The qualitative research methodology includes the following.
1. In this study, the disease in focus is “breast cancer.”
2. The drugs chosen are of the “targeted therapy” type.
3. The population or market of interest is the United States of America along with Europe for comparison purposes.
The review work was carried out by thorough searching of the following secondary sources of information.
a. Official websites (FDA site, Clinicaltrial.gov, cancer. gov, and so on). Table 1 shows health authorities of chosen countries.
b. Commercial websites (pharma company websites, white paper, and fierce pharma site).
c. Annual reports.
d. Articles/literature (Scopus/PubMed).
The criteria for selection of breast cancer drugs for the study are
a. If the drug is in current use.
b. If the drug is a blockbuster or has a market value (in $)—a medicine that is incredibly popular and brings in at least $1 billion in revenue annually for the firm that sells it is known as a blockbuster.
c. If it belongs to a targeted therapy group.
To find time period for each drug’s approval by finding the starting date of new drug application (NDA) filing and approval date of selected drugs and comparing their timelines.
To identify regulatory challenges faced during clinical trial and approval phases. This is done by going through some articles collected from Scopus, PubMed, Web of Science, Science Direct, and so on, news items, and also annual reports of companies to which selected drugs belong. To propose a solution for the identified causes of delay in the approval of breast cancer drugs.
 | Table 1. The health authorities of chosen countries. [Click here to view] |
RESULT
A NDA “tells the whole narrative of a medicine,” according to the FDA. A comprehensive application, therefore, contains all information about the drug, from preclinical studies to Phase 3 clinical trials. The FDA has 60 days from the time an NDA is submitted to decide whether to accept the application for review or reject it. The Center for Drug Evaluation and Research will respond within 6–10 months if it is submitted for review. If the FDA deems that the merits of the drug surpass the risks, they shall approve an NDA. If a drug is not authorized, the FDA will alert the candidate and sponsor in writing. This could be a major setback, thus it is crucial to submit an NDA that is factual, thorough, and well-written [12].
The duration for approval of selected drugs in the USA and Europe was calculated from the time of NDA filing to approval and is tabulated in Table 2.
DISCUSSION
Based on the popularity of the companies and the criteria matched by various drugs, the following were selected for the study: abemaciclib, palbociclib, pertuzumab, and lapatinib.
Since EMA has a different approval pathway, there is an “approval lag” in the timelines, which is evident in Table 2. Hence, there is a need to harmonize the regulations of breast cancer clinical trials and the costs involved to speed up the process of NDA approvals. There is a need for inclusive collaboration between reference nations such as the US and Europe to avoid the approval lag of potent drugs for patients.
Regulatory challenges during breast cancer clinical trial and approval
Clinical trials are the sole phase that is absolutely necessary for bringing novel drugs to market and creating value. Clinical testing can cost up to $2.6 billion per medicine, according to a 2014 study by the Tufts Center for the Study of Drug Development. In less than 20 years, the price of developing new drugs has increased by 400% [37].
Regulatory barriers and approval delays
Meeting compliance responsibilities is understandably one of the main obstacles preventing the timely and cost-effective completion of clinical trials, given how strictly regulated the pharmaceutical sector is. Trials are becoming limited by their own complexity, particularly as they spread internationally. It is getting increasingly difficult to coordinate between numerous sites, partners, and vendors. If data is not properly saved and organized, even simple actions like version control on consent form documents might result in serious protocol violations and regulatory disasters. In actuality, maintaining trial compliance consumes more than one-third of clinical research spending. Using software systems helps greatly. Record-keeping systems assist in keeping tabs on deadlines for ethics/IRB (Institutional review board) submissions and approvals after a trial is underway, assisting in maintaining study timeframes [37].
Site selection and recruitment
One of the main causes of delays and trial failures is the inability to find and keep enough participants to complete a trial. Patient access, infrastructure, and suitability for the specified therapy type are some of the most crucial criteria for site selection, which is a crucial first stage in the patient recruitment process. Pharma companies have the resources to locate available investigators with the greatest potential for enrollment. There are more variables, relationships, and possible outcomes to take into account as trial complexity rises. Due to its intricacy, it may be challenging to weigh all the repercussions of a choice, which could result in mistakes and oversights. Due to the variety of study sites, geographic heterogeneity brings new complications. Decision-making may become more complicated and demanding depending on the legislation, cultural considerations, and logistical issues that must be managed in a given place. The unpredictable nature of trials depends critically on the rate of change. Decisions must be made on the spot due to the trial procedure being heavily impacted by rapidly changing conditions and new data. Effective decision-making techniques and rapid thinking are necessary to keep up with the pace of change. Roles and responsibilities among staff members change often, and dealing with diverse data sources and a lack of data visibility makes it difficult to react swiftly. Artificial intelligence can use operational data from prior studies to forecast future site performance. In the future, it might even be able to forecast retention, trial success, and if a medicine will provide favorable results [37].
Surrogate endpoints versus event endpoints
Anti-breast cancer therapies attempt to prolong patients’ overall survival or elevate their standard of living. Surrogate endpoints are frequently used in oncology studies since they may be assessed more quickly than event endpoints, which frequently necessitates lengthier research. Accelerated FDA approval is another obvious advantage. Surrogacy should, however, be carefully considered in the experimental phases of pharmaceuticals. Surrogate endpoints derived by extrapolation may not be taken as accurate measures of a drug’s effectiveness, particularly if no subsequent research can confirm the strength of the surrogate-survival correlation [38].
Applying for accelerated approval
The FDA will approve a medicine and permit it to be marketed in the United States if it determines that the advantages of the drug exceed the known hazards. However, the FDA may send a detailed response letter if an NDA has issues or if further information is required to make that decision. In accordance with accelerated approval regulations (21 CFR part 314, subpart H and 21 CFR part 601, subpart E), the FDA may revoke the approval if studies do not support the preliminary findings. Unexpected safety risks that arise or a failure to prove a drug’s efficacy are frequent issues mentioned in most full answer letters. A sponsor could need to carry out additional research, possible ones with more participants, studies with various participant kinds, or trials lasting longer. A formal NDA presubmission meeting should be requested by applicants 4–6 months before the anticipated submission date, according to the FDA. Presubmission meetings are optional, but they offer a great chance to get FDA input and ask questions before submission. These presubmission procedures draw attention to the crucial role that regular communication with the FDA plays in an effective NDA [39,40].
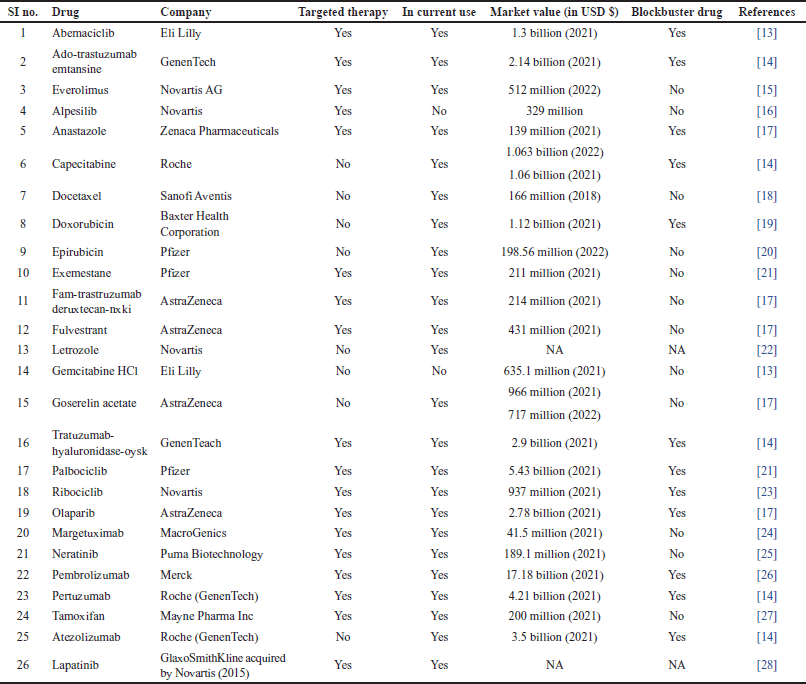 | Table 2. Criteria for selection of drugs. [Click here to view] |
Using Prescription Drug User Fee Act (PDUFA) by companies
More than 1,000 novel pharmaceuticals and biologics have entered the market since PDUFA was approved in 1992, including treatments for cancer, AIDS, cardiovascular disease, and life-threatening infections. PDUFA has enabled the FDA to continue the same thorough review process while bringing access to new pharmaceuticals as quickly as or faster than anyplace else in the world. In accordance with PDUFA, pharmaceutical firms agree to pay fees that increase FDA resources, and the FDA accepts deadlines for reviewing NDAs. Drug user fees assist the FDA in upgrading its information technology resources in addition to funding more personnel. By accepting more electronic applications and storing review materials electronically, the agency has shifted toward an electronic submission and review environment. The 2002 amendments to PDUFA set a 10-month goal for a standard review [39].
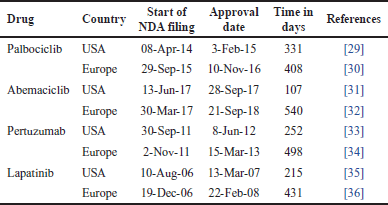 | Table 3. Approval timelines of selected drugs. [Click here to view] |
Absence of scientific knowledge of new breast cancer therapies
It is challenging to determine the likelihood of safety and efficacy for new therapies because of their nature, which has yet to show the full amount of its power (or lack thereof) over a wide range of human factors. Oncology clinical trials, especially breast cancer trials, have a hit-or-miss quality due to the lack of preclinical data and postmarketing reports, which can significantly raise the cost of the program and raise the probability of failure in Phase 3. The effectiveness and toxicity levels of molecularly targeted agents can take years to establish, whereas the good or negative impacts of chemotherapy treatments are often seen during active treatment. This alone might have a significant effect on recruitment because patients could be hesitant to sign up for a program with too much uncertainty and no sufficient data [38].
Misuse of data and statistics
The statistics should not be used if they are unimportant or lack scientific support. They might introduce errors into the patient recruitment procedure and prevent them from really reaching the individuals who would gain the most from novel cancer treatment. Statistics are increasingly used in reports and academic papers to support hypotheses. But one must approach cautiously, especially if no biostatisticians are present in the team. Throughout the course of breast cancer clinical trials, utilization of the proper software to manage the data reduces the possibility of human mistakes, misunderstanding, and misuse [38].
Usage of local language for submissions
Breast cancer trials are becoming multicentric. They are spread across multinations and occur simultaneously to meet the recruitment goals as well as test diverse populations. To make patients understand the benefits and risks of the drug trial, the usage of local language is better. Not only patients, but the personnel involved in trials also find it useful if the documents are handled in their local languages (trial protocol, investigator’s brochure, and so on). It may be reasonable to use local languages for some study documents submitted to study sites and IRBs because these documents are utilized by many local users. The use of local language for submissions is crucial from a regulatory standpoint as approvals can happen more quickly if local authorities can assess submissions in their native tongue. The difficulty comes up when translating documents from English into regional languages. Meaning may be skewed, and the company will have to submit more information and respond to more inquiries from the health authority, which would delay approval. It also has an impact on the company’s finances because translating large documents costs a lot of money.
Data management
One of the top tasks of trial management during active trials is making sure that locations are closely monitored, and data is being recorded correctly. It takes active data analysis and monitoring to track compliance, including monitoring endpoint data, deviations, and any adverse events, to ensure patient perseverance and completion of the clinical trial protocol. When done manually, as many pharmaceutical businesses still do, it is challenging to swiftly update information and aggregate data from various sources and across various IT platforms. Data management software provides the advantage of centralizing information promptly and making it easily available to critical individuals. The proper system can seamlessly monitor protocol compliance while integrating across many data sources in close to real-time, enabling proactive reaction to severe adverse effects or adverse effects . In addition, data managers can automate the reporting of results, making it simple to spot patterns in patients’ answers earlier [37].
Overcoming unique drug safety and regulatory challenges
Typically, businesses lack the resources and knowledge necessary to set up an internal pharmacovigilance (PV) operation because doing so would take away crucial time and funds from product development. As a result, non-PV-specific organizations involved in clinical development or regulatory supervision frequently seize control. After product approval, PV takes center stage, requiring motivated, qualified employees to manage operations and support systems. To carry out tasks such as safety reporting, benefit-risk analysis, signal identification, and risk management strategies, highly experienced personnel must be kept. Standard operating procedures and safety management techniques are necessary for regulatory compliance. Missing deadlines, rework, poor-quality output, inefficient procedures, and noncompliance problems can increase expenses. Warning letters are sent by regulatory agencies for infractions, and there may be serious repercussions. It is expensive and time-consuming to design procedures for all phases of clinical development, regulatory, and PV activities. Organizations that specialize in outsourcing can offer reliable, audited systems, saving time and money. These procedures can be tailored to the needs of the business and are frequently updated to take into account evolving legal requirements and technological developments [41].
Postapproval change management
Although a crucial component of life cycle management, it faces regulatory obstacles. Two causes can be linked to delays in global approvals brought on by protracted CMC (Chemistry Manufacturing Control) postapproval regulatory processes: problems with dossier preparation and problems with data. These challenges arise as a result of obsolete legislative frameworks and the inability to utilize cutting-edge technologies for the sharing of information, including data and narratives. The high workload and duplication in postapproval regulatory submissions are caused by a number of issues, including the dispersion of data across numerous document sections, regional variations, staggered filing schedules across markets, and the enormous volume of data locked in PDF format. The buildup of these redundancies hinders innovation and causes delays in the deployment of crucial postapproval CMC (Chemistry Manufacturing Control) improvements [42].
CONCLUSION
The timeline for approval of these drugs was found by calculating the duration between the initial NDA filing date and the final approval date given by the US FDA and EMA. The startling differences between the US FDA and EMA’s approval times showed that these two authorities, despite being well-established and adhering to strict standards of safety, efficacy, and quality of drugs, needed to continue harmonizing their procedures. This process has increased cooperation and information sharing between the two organizations, but it has not yet encouraged the exchange of opinions.
To provide quicker access to anticancer medications, the FDA is more willing to take risks and grant approval based on less reliable data, though it does so more easily than the EMA. The understanding from achieving the first objective is that harmonization of regulatory requirements in different countries can be beneficial in terms of reducing duplication of efforts by pharmaceutical companies, increasing efficiency, improving access to innovative treatments, and facilitating global trials which is very much needed in breast cancer drug studies.
Based on the study conducted to map the regulatory challenges in breast cancer products’ lifecycle, it can be concluded that there are several challenges that need to be addressed. It was found that the regulatory challenges vary depending on the product’s lifecycle and they can arise from different regulatory bodies and stakeholders involved. By understanding these challenges, companies can further identify potential barriers to their particular product and develop strategies to overcome them based on the market they will be targeting. Certain solutions to avoid delay in the approval of innovative breast cancer therapies are the usage of accelerated approval pathways by having predefined surrogate endpoints, by using different schemes such as PDUFA, by taking the help of Contract Research Organizations especially when the trials are multinational and multicentric and by using modern software for better handling of data and maintain compliance as all processes are moving from paper-based format to electronic form. One such software solution is the Regulatory Information Management system.
Findings from this study may inform future regulatory strategies and interventions aimed at addressing these challenges and improving the overall regulatory process of breast cancer products ultimately contributing to the advancement of breast cancer care and patient outcomes. Further research and collaboration among stakeholders are needed to overcome these challenges and ensure that breast cancer products are safe, effective, and accessible to those who need them.
ACKNOWLEDGMENT
The authors would like to express their sincere gratitude to all the individuals and organizations that have contributed to the publication of this review article.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJEs) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICT OF INTEREST
The authors have no conflicts of interest regarding this investigation.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. National Breast Cancer Foundation. Breast cancer facts & figures 2022-2024 [Internet]. [cited 2023 Apr 12]. Available from: https://www.nationalbreastcancer.org/breast-cancer-facts/
2. National Breast Cancer Foundation. Breast cancer facts & stats [Internet]. [cited 2022 Dec 12]. Available from: https://www.nationalbreastcancer.org/breast-cancer-facts/
3. National Breast Cancer Foundation. Breast cancer treatment options [Internet]. [cited 2023 Apr 12]. Available from: https://www.nationalbreastcancer.org/breast-cancer-treatment/
4. National Cancer Institute. Targeted therapy to treat cancer [Internet]. [cited 2022 Dec 13]. Available from: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies
5. Hein T. Product lifecycle management for the pharmaceutical industry [Internet]. [cited 2022 Dec 15]. Bethesda, MD: ISPE. Available from: https://pharmaceutical.report/Resources/Whitepapers/f09463b8-48a6-4bfc-b3cd-6df45bb110af_lifecycle-mgmt-pharmaceutical-bwp-070014.pdf
6. Eby K. Data, data, everywhere! Product lifecycle management in the world of IoT [Internet]. [cited 2023 Apr 12]. Available from: https://www.smartsheet.com/product-life-cycle-management
7. Investopedia. Product life cycle explained: stage and examples [Internet]. [cited 2023 Jan 1]. Available from: https://www.investopedia.com/terms/p/product-life-cycle.asp
8. AZ Regulatory. Intelligence plus health authority requirements = sound strategy [Internet]. [cited 2023 Feb 23]. Available from: https://www.az-regulatory.com/services/strategy
9. Trilogy Writing & Consulting. Cutting back: solutions to reduce the time and costs of clinical development [Internet]. [cited 2023 Apr 12]. Available from: https://trilogywriting.com/document/cutting-bacl-solutions-reduce-time-costs-clinical-development/
10. US Food & Drug Administration [Internet]. [cited 2023 Feb 12]. Available from: https://www.fda.gov/
11. European Medicines Agency [Internet]. [cited 2023 Feb 12]. Available from: https://www.ema.europa.eu/en
12. Worldwide Clinical Trials. NDA Submission 101: anticipating study requirements early [Internet]. [cited 2023 Apr 13]. Available from: https://www.worldwide.com/blog/2020/05/nda-submission-101-anticipating-study-requirements-early/
13. Statista. Eli Lilly top products by revenue 2021-2022 | Statista [Online]. New York, NY: Statista; 2022 [cited 2023 Jan 09]. Available from: https://www.statista.com/statistics/266529/pharmaceutical-company-roche-top-drugs-based-on-revenue/
14. Roche. ar21e.pdf [Internet]. Basel, Switzerland: Roche; 2022 [cited 2023 Jan 9]. Available from: https://www.roche.com/investors/reports-results/reports/2021/ar21e.pdf
15. Novartis. Product sales | Novartis [Internet]. Basel, Switzerland: Novartis; 2022 [cited 2023 Jan 9]. Available from: https://www.novartis.com/our-focus/product-sales
16. Novartis. Novartis annual report 2021 [Internet]. Basel, Switzerland: Novartis; 2021 [cited 2023 Jan 9]. Available from: https://www.novartis.com/sites/www.novartis.com/files/novartis-annual-report-2021-en.pdf
17. AstraZeneca. AstraZeneca_AR_2021.pdf [Internet]. Cambridge, UK: AstraZeneca; 2021 [cited 2023 Jan 9]. Available from: https://www.astrazeneca.com/content/dam/az/PDF/2021/annual-report-and-form-20-f-2021.pdf
18. Sanofi. 2021 Sanofi 20-F [Internet]. Paris, France; Sanofi; 2021 [cited 2023 Jan 9]. Available from: https://www.sanofi.com/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/common/docs/investors/2021/sanofi-2021-20-f.pdf
19. PR Newswire. Global doxorubicin market report 2022: market to reach $1.3 billion by 2026—competition, new entrants seek opportunities, recent market activity [Internet]. [cited 2023 Jan 9]. Available from: https://www.prnewswire.com/news-releases/global-doxorubicin-market-report-2022-market-to-reach-1-3-billion-by-2026---competition-new-entrants-seek-opportunities-recent-market-activity-301509505.html
20. GlobeNewswire. The global epirubicin market is expected to reach 243.99 [Internet]. [cited 2023 Jan 9]. Available from: https://www.globenewswire.com/news-release/2022/04/26/2443063/0/en/The-global-epirubicin-market-is-expected-to-reach-243-99-million-by-2026-at-a-CAGR-of-6-0-from-2022-to-2026.html
21. Pfizer. Pfizer Inc. 2022 form 10-K [Internet] New York, NY: Pfizer; 2022 [cited 2023 Jan 10]. Available from: https://s28.q4cdn.com/781576035/files/doc_financials/2022/ar/PFE-2022-Form-10K-FINAL-(without-Exhibits).pdf
22. Novartis. Product sales [Internet]. Basel, Switzerland: Novartis [cited 2023 May 01]. Available from: https://www.novartis.com/investors/financial-data/product-sales
23. Novartis. Novartis announces strong Q4 and full year 2021 results, marking progress on portfolio transformation [Internet]. Basel, Switzerland: Novartis; 2022 [cited 2023 Jan 10]. Available from: https://www.novartis.com/sites/novartis_com/files/q4-2021-media-release-en.pdf
24. GlobeNewswire. MacroGenics provides update on corporate progress and first quarter 2021 financial results [Internet]. GlobeNewswire; 2021 Apr 29 [cited 2023 Jan 10]. Available from: https://www.globenewswire.com/en/news-release/2021/04/29/2220145/0/en/MacroGenics-Provides-Update-on-Corporate-Progress-and-First-Quarter-2021-Financial-Results.html
25. Puma Biotechnology. 2021 annual report [Online]. Los Angeles, CA: Puma Biotechnology; 2021 [cited 2023 Jan 10]. Available from: https://s24.q4cdn.com/201644000/files/doc_financials/2021/ar/042922-Puma-2021-AR-Final-bkmk.pdf
26. Merck. Merck announces fourth-quarter and full-year 2021 financial results. Rahway, NJ: Merck; 2022 Feb 8. Available from: https://www.merck.com/news/merck-announces-fourth-quarter-and-full-year-2021-financial-results/
27. Maximize Market Research. Tamoxifen citrate market size, share | industry report, 2027 [Internet]. [cited 2023 Jan 11]. Available from: https://www.maximizemarketresearch.com/market-report/tamoxifen-citrate-market/148744/#:~:text=Tamoxifen%20Citrate%20Market%20size%20was%20valued%20at%20US%24,2022%20to%202027%2C%20reaching%20nearly%20US%24%20232.91%20Mn
28. Novartis. Novartis announces completion of transactions with GSK [Internet]. Basel, Switzerland: Novartis; 2015 [cited 2023 May 01]. Available from: https://www.novartis.com/news/media-releases/novartis-announces-completion-transactions-gsk
29. US Food and Drug Administration. FDA approves Ibrance for postmenopausal women with advanced breast cancer [Press release]. 2015 Feb 3. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-ibrance-postmenopausal-women-advanced-breast-cancer
30. European Medicines Agency. First breast cancer medicine in 10 years backed by EMA. Amsterdam, Netherlands: EMA; 2021 Sep 29. Available from: https://www.ema.europa.eu/en/news/first-breast-cancer-medicine-10-years-backed-ema
31. FDA. FDA approves new treatment for advanced breast cancer. Silver Spring, MD: FDA; 2019 Nov 15 [cited 2023 Mar 15]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-advanced-breast-cancer
32. European Medicines Agency. Abemaciclib [Internet]. Amsterdam, Netherlands: EMA [cited 2023 Mar 10]. Available from: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/abemaciclib-0
33. Genentech. Genentech submits new drug application for trastuzumab-DM1 in previously treated metastatic HER2-positive breast cancer [Internet]. 2011 Sep 30 [cited 2023 Mar 10]. Available from: https://www.gene.com/media/press-releases/12646/2011-09-30/genentech-submits-new-drug-application-for-trastuzumab-dm1-in-previously-treated-metastatic-her2-positive-breast-cancer
34. EMA. Perjeta-EPAR-public assessment report [Internet]. Amsterdam, Netherlands: EMA; 2013. Available from: https://www.ema.europa.eu/en/documents/assessment-report/perjeta-epar-public-assessment-report_en.pdf
35. FDA. NDA 22-059 [Internet]. Silver Spring, MD: FDA [cited 2023 Mar 12]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/022059s000ltr.pdf
36. European Medicines Agency. Assessment report For tyverb [Internet]. Amsterdam, Netherlands: EMA [cited 2023 Mar 12]. Available from: https://www.ema.europa.eu/en/documents/assessment-report/tyverb-epar-public-assessment-report_en.pdf
37. P360. 4 Challenges with managing clinical trials & how technology can help [Internet]. 2023 [cited 2023 Apr 12]. Available from: https://www.p360.com/trials360/4-challenges-with-managing-clinical-trials-how-technology-can-help/
38. ProPharma Group. 6 Unique challenges hindering oncology clinical trials [Internet]. 2023 [cited 2023 Apr 12]. Available from: https://www.propharmagroup.com/thought-leadership/6-unique-challenges-hindering-oncology-clinical-trials
39. FDA. FDA’s drug review process: continued [Internet]. Silver Spring, MD: FDA [cited 2023 Apr 12]. Available from: https://www.fda.gov/drugs/information-consumers-and-patients-drugs/fdas-drug-review-process-continued
40. Worldwide Clinical Trials. NDA Submission 101: anticipating study requirements early. 2020 May 12. Available from: https://www.worldwide.com/blog/2020/05/nda-submission-101-anticipating-study-requirements-early/
41. Safety & regulatory solutions for small and medium-sized life science organisations [Internet]. [cited 2023 Apr 12]. Available from: https://drugdevelopment.labcorp.com/content/dam/covance/assetLibrary/whitepapers/Safety-and-Regulatory-WPCMA010.pdf
42. ISPE. Streamlining postapproval submissions using ICH Q12 & SCDM [Internet]. Bethesda, MD: ISPE [cited 2023 Apr 12]. Available from: https://ispe.org/pharmaceutical-engineering/september-october-2022/streamlining-postapproval-submissions-using-ich#:~:text=Delays%20in%20global%20approvals%20experienced%20due%20to%20lengthy,challenges%20with%20dossier%20preparation%20and%20challenges%20with%20data