
INTRODUCTION
Depression is a clinical syndrome associated with the alteration of a person’s mood. It is estimated that about 280 million people which comprise 3.8% of the world population are affected by this illness. Approximately 5% of adults and 5.7% of the elderly suffer from depression. According to the Journal of the American Medical Association, women are more prone to depression than men. Depression is said to cause suicidal tendencies, leading to around 700,000 deaths yearly, accounting for the fourth major cause of death in 15 to 29 years old (Depression, n.d.; Evans-Lacko et al., 2018).
The major symptoms of depressive disorder include lack of interest, sadness, fatigue, fluctuations in weight, altered sleep patterns, irritation, lowered self-esteem, and suicidal notions. Depression is said to develop due to complex social, biological, and psychological factors like stress, adverse life events, genetics, alterations in the biochemical aspects of the body, physical health, and so on (Brigitta, 2002; Mathew et al., 1981). Although several known risk factors contribute to the illness, the exact etiology leading to the disorder is indefinite. Based on the symptoms, severity, and pattern of occurrence diagnostic and statistical manual of mental disorders classifies depression into unipolar (single episode) and bipolar (recurrent) (Kendell, 1976), whereas the ICD-10 classification of mental and behavioral disorders divides it into three categories, namely mild, moderate, and severe (Paykel, 2008).
Mild depression itself might not call for medical attention, but a persistently depressed state especially if it is accompanied by other symptoms could signal the presence of a serious medical or psychiatric condition that needs to be treated. When treating depression, a combination of psychotherapy and antidepressants is frequently used. The majority of the time, tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRI), reversible inhibitors of MAO-A, and serotonin and noradrenaline reuptake inhibitors (SSNRI) are employed (Duval et al., 2006; Givens et al., 2007). In addition to the conventional antidepressants’ effectiveness, medication therapy may have unintended consequences such as serotonin toxicity, hyperglycemia, nausea, weight gain, dizziness, tremors, altered sexual functionality, drug interactions, and drug-induced QT prolongation. Due to the frequent dose schedule, it is also discovered that drug adherence is a significant issue. This encourages the development of novel therapeutics with reduced toxicity and enhanced activity (Cascade et al., 2009; Kelly et al., 2008).
Serotonin reuptake is the process by which the neurotransmitter serotonin is transported from the synaptic cleft back to the presynaptic neuron by the serotonin transporter (SERT or 5-HTT), which stops the action of serotonin and recycles it in a sodium-dependent manner. Many SSRIs and tricyclic antidepressants, and other antidepressants reduce serotonin reuptake via binding to SERT (Bank, n.d.; Owens and Nemeroff, 1994; Squire, 2008). 5-HT2A is another such receptor belonging to the serotonin receptor family which is known to be involved in the action of several antipsychotic medicines mainly in the management of bipolar disorders and mood stabilization. Patients who attempted suicide or were depressed had elevated 5-HT2A receptors than healthy individuals. These findings imply that the pathophysiology of depression involves post-synaptic 5-HT2A over-density. SSRIs and atypical antipsychotics are known to follow the mechanism that causes chronic downregulation of the post-synaptic 5-HT2A receptor (Bank, n.d.; Eison and Mullins, 1995; Martin et al., 1998).
Naturally occurring heterocyclic compounds are actively being researched for their potential role in the development of innovative therapeutic agents in the current era of developing diseases. Among a large group of these heterocyclic compounds, thiophene and oxazepines are the classes of molecules that caught the attention of medicinal chemists, leading to the development of their vast therapeutic value as an anti-inflammatory, antipsychotic, analgesic, and antimicrobial agents. (Berrade et al., 2011; Gibbs et al., 1976; Shah and Verma, 2018; Sharma et al., 2008; Wardakhan et al., 2008) resulting in encouraging findings that spur the creation of new therapeutic compounds.
Many tricyclic antidepressant medications, including Amoxapine, Sintamil, and others, contain the heterocyclic nucleus oxazepine, and Duloxetine, an SSNRI used to treat depression and anxiety, contains a thiophene nucleus, demonstrating the possibility of the same being an effective treatment for depression and anxiety. Several studies present the possible activity of the thiophene molecule as an antidepressant agent which sparked the notion of creating a single hybrid molecule containing both of these moieties and evaluating their biological activity.
MATERIALS AND METHODS
In silico platform
Using Maestro 12.3 version programmed on DELL Inc.27” workstation on Intel Core i7-7700 CPU@ 3.60 GHz ×8 processor with 1,000 GB hard disk and 8 GB RAM, all the in-silico analysis was performed. The operating system used was Linux –×86_64. (Schrödinger | Schrödinger is the scientific leader in developing state-of-the-art chemical simulation software for use in pharmaceutical, biotechnology, and materials research., n.d.)
Molecular docking
Using ChemDraw 20.1.1 application, the 2D structures as given in Table 10 and Canonical SMILES of the ligand compounds were obtained which were converted into 3D images using the Ligprep module on the Schrödinger. The imported ligands were energy minimized. The proteins 5-HT2AR (PDB ID: 6WGT) and 5-HTT (PDB ID: 7LIA) obtained from the protein data bank (https://www.rcsb.org/) were preprocessed, refined, optimized, and minimized using the protein preparation wizard of Schrödinger. The protein’s active site was identified and a grid was generated using the Grid Generation module. Finally, using Glide extra precision scoring tools, the protein and ligands were docked. The docking scores were compared with a standard antidepressant drug Imipramine (Dwivedi et al., 2021; Kalirajan et al., 2019; Mathew et al., 2014).
Prime molecular mechanics-generalized born surface area (MM-GBSA) binding free energy
The MM-GBSA method was employed to calculate the binding energy of the receptor-ligand complex. By taking into account the solvation model for polar and non-polar solvation as well as molecular mechanics energies, Schrodinger’s Prime module determined the ΔGbind in kcal/mol (Genheden and Ryde, 2015; Wang et al., 2019).
Pharmacophore modeling
Using the phase module of Schrödinger, pharmacophore models of top-score compounds were generated. Multiple ligand method was used for the same. In reference to virtual screening on huge chemical databases, the term “pharmacophore” refers to a collection of steric and electronic characteristics that validate the best supramolecular interactions. It describes the 3D configurations of functional groups necessary for biological action. It is a more effective and potent method for finding compounds that may be used to either stimulate or inhibit macromolecular activity.
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) and physicochemical properties
Using the QikProp module of Schrödinger, the ADMET and physicochemical parameters of the ligand molecules were predicted. Validation of the rule of five was also carried out using the QikProp module (Duffy and Jorgensen, 2000; QikProp User Manual, n.d.). Absorption, distribution, metabolism, and excretion (ADME) of the administered molecule play an important role in the bioactivity of a molecule. Lipinski’s rule of five helps in predicting the oral bioavailability of any molecule.
Molecular dynamic (MD) simulation
The MD Simulations were conducted using Gromacs version 2023.1. To prepare the protein topology, the charmm27 all-atom forcefield was employed with the pdb2gmx module of Gromacs. The proteins were immersed in a dodecahedron box with dimensions of 1 nm in all directions, using a 3-point water model for solvation. Sodium (Na+) and Chloride (Cl−) ions were added as counter ions to neutralize the system. The energy minimization process utilized the steepest descent integrator and a verlet cutoff scheme for a maximum of 50,000 steps, aiming to achieve the lowest energy confirmation. The system was then equilibrated for 100 picoseconds using Canonical [constant number (N), constant volume (V), and constant temperature (T)] and Isobaric [constant number (N), constant pressure (P), constant temperature (T)] ensembles. The thermostat V-rescale was employed to maintain a constant temperature of 300 K, while the C-rescale pressure-coupling algorithm was used to maintain a constant pressure of 1 bar. For long-range electrostatics, coulomb, and van der Waals calculations, the Particle Mesh Ewald method was applied with a cutoff of 1.2 nm. The LINCS algorithm was utilized to constrain bond lengths. The MD run was conducted for 100 ns, with coordinates and energies saved at every 10 picoseconds. The resulting trajectories were analyzed using the built-in utilities provided by Gromacs. After the MD run, various parameters including root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG), and solvent accessible surface area (SASA) were evaluated. Additionally, the University of California, San Francisco Chimera Visualizer was employed to visualize the complex at the beginning and end of the MD run (Dwivedi et al., 2021; Kalirajan et al., 2019; Mathew et al., 2014).
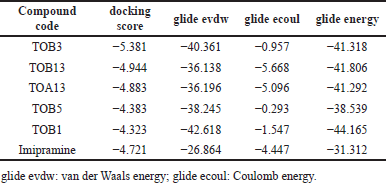 | Table 1. Molecular docking scores of in silico potential compounds with protein 7LIA. [Click here to view] |
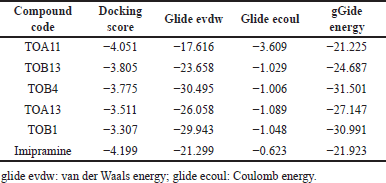 | Table 2. Molecular docking scores of in silico potential compounds with protein 6WGT. [Click here to view] |
RESULTS AND DISCUSSION
Molecular docking
The ligand molecules TOB3, TOB13, and TOA13 with docking scores −5.381, −4.944, and −4.883 kcal/mol, respectively, docked against the protein 5-HTT exhibited better scores than the standard Imipramine (−4.721 kcal/mol). The docking scores of other compounds ranged from −2.00 to −4.383. On the other hand, the 5-HT2AR inhibitory activity of the ligands extended from −1.696 to −4.051. The compound TOA11 exhibited an appreciable score (−4.051 kcal/mol) compared to the standard Imipramine (−4.199 kcal/mol). The van der Waals energy, coulomb energy, docking energy, and the interactions of each ligand with functional residues of the proteins are given in Tables 1–4 and Figures 1–4.
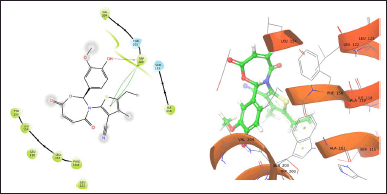
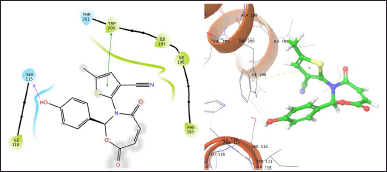
The molecule TOB3 showed hydrophobic interaction with amino acid residues Ile108, Ala331, Phe335, Tyr495, Pro499, Phe556, Pro560, and Pro56. The polar interaction with Gln332, Ser559, Thr497, hydrogen bonding with Thr497, whereas with Arg104 and Asp328, Glu494 exhibited positive and negative interactions, respectively, while docked against the 5-HTT protein. The ligand TOB13 was observed to exhibit hydrophobic interactions with Ile108, Ala331, Phe335, Pro499, Phe556, Leu563, Pro561, polar interactions with Gln332, Thr497, Ser555, Ser559, Gln562; positive and negative interactions with Arg104 and Asp328, Glu494, respectively, along with hydrogen bonding with Thr497 residue. The molecule TOA13 also depicted hydrophobic (Ala331, Ile327, Pro560, Pro561, Leu557, Phe556, Pro499, Tyr495, Tyr579), polar (Thr497, Ser559, Gln562), hydrogen bonding (Glu494), positive (Arg104), and negative (Asp328, Glu494) interactions followed by the pi-pi stacking (Phe556) with different amino acid residues. Docking of molecules against the protein 5-HT2AR yielded appreciable results. The ligand TOA11 showed hydrophobic (Ile118, Phe193, Ile196, Ile197, Trp200) and polar (Ser115, Thr201) interactions, hydrogen bonding (Ser115) and pi-pi stacking (Trp200) with several amino acid residues of the protein.
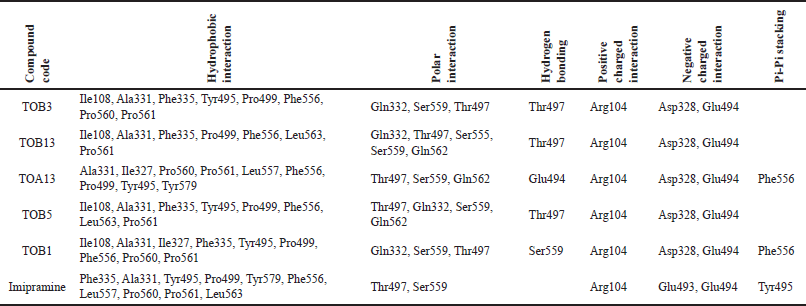 | Table 3. Molecular docking interactions of in silico potential compounds with the protein 7LIA. [Click here to view] |
 | Table 4. Molecular docking interactions of in silico potential compounds with the protein 6WGT. [Click here to view] |
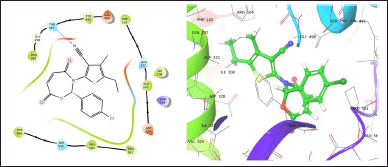 | Figure 1. TOB3 with 7LIA. [Click here to view] |
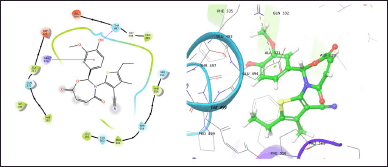 | Figure 2. TOB13 with 7LIA. [Click here to view] |
 | Figure 3. TOA11 with 6WGJ. [Click here to view] |
 | Figure 4. TOB13 with 6WGJ. [Click here to view] |
Binding free energy calculation
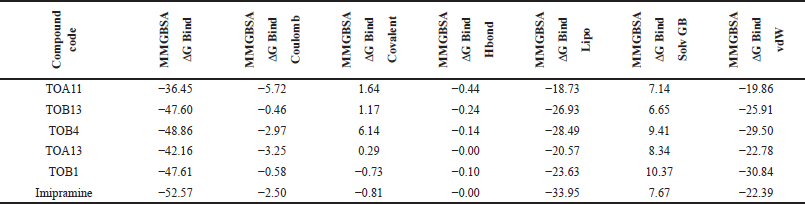
The docking results were validated using binding free energy analysis of the protein-ligand complexes. The ΔGbind of the protein 7LIA complexed with different ligands was in the range of −11.58 to −56.39 kcal/mol, whereas the complexes with the protein 6WGT ranged from −31.06 to −57.75 kcal/mol (Tables 5 and 6).
Pharmacophore modeling
To identify the chemical features of the best-interacted molecules TOB3 (with the protein 7LIA) and TOA11 (with the protein 6WGT) that are responsible for the interaction with the active site of the protein, pharmacophore modeling was employed (Fig. 5). Acceptors (A4, A2, A5), aromatic ring (R11), and hydrophobic interactions (H8) were found to contribute to the interaction in the case of molecule TOB3. Whereas acceptor (A3), donor (D6), aromatic rings (R8, R9), and hydrophobic interaction (H7) were the attributes responsible for the molecule TOA11. The acceptors present in the compound TOB3 are separated by the distance 2.25 (A2–A4), 4.07 (A2–A5), and 5.29 (A4–A5) forming a pharmacophoric triangle with corresponding angles of 110.1°, 23.6°, and 46.3°, respectively. The aromatic rings in the molecule TOA11 were found to be at a distance of 4.05 (R8–R9) with an angle of 43.5° (∠R8R9H7).
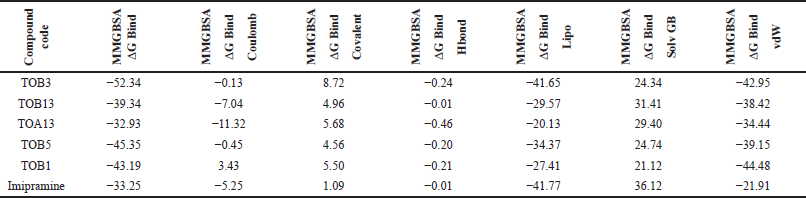 | Table 5. Binding free energy calculation of in silico potential compounds with protein 7LIA. [Click here to view] |
 | Table 6. Binding free energy calculation of in silico potential compounds with protein 6WGT. [Click here to view] |
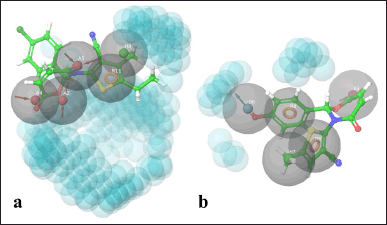 | Figure 5. Pharmacophore modeling. [Click here to view] |
Predicted ADME profile
Among the tested molecules for their ADME profile, the compounds except for TOA1, TOA2, TOA10, TOA11, TOA13, TOB1, TOB2, TOB11, and TOB13 were predicted to have a high percentage of human oral absorption. The predicted apparent Caco-2 cell permeability of the compounds was found to be moderate ranging from 42.011 (TOB2) to 477.045 nm/second (TOB3). It was also observed that the molecules could moderately bind to the protein human serum albumin. The compounds’ predicted brain/blood partition coefficient was also within the recommended range of −3 to 1.2. The compounds TOA1, TOA2, TOA11, TOA12, TOA13, TOB1, TOB2, TOB10, TOB11, TOB12, and TOB13 were predicted to be CNS inactive whereas the molecules TOA6, TOA7, TOB3, TOB4, TOB5, TOB6, and TOB7 were found to be moderately active. The predicted number of likely metabolic reactions of all the compounds was also found to be within the prescribed range (Table 7).
Predicted solvent-accessible surface area
A biomolecule’s surface area accessible to the solvent is termed SASA and has a recommended range from 300 to 1,000. All the ligand molecules involved in the study were found to be within the prescribed limit. The molecules were also checked for FOSA (Hydrophobic component of SASA with a recommended range of 0–750), FISA (Hydrophilic component of SASA having a recommended range of 7.0–330), PISA (Pi component of SASA with a recommended range of 0–450), and total solvent-accessible volume in cubic angstroms using a probe with a 1.4 Å radius (Recommended range: 500–2000). It was observed that the compounds were within the suggested range (Table 8).
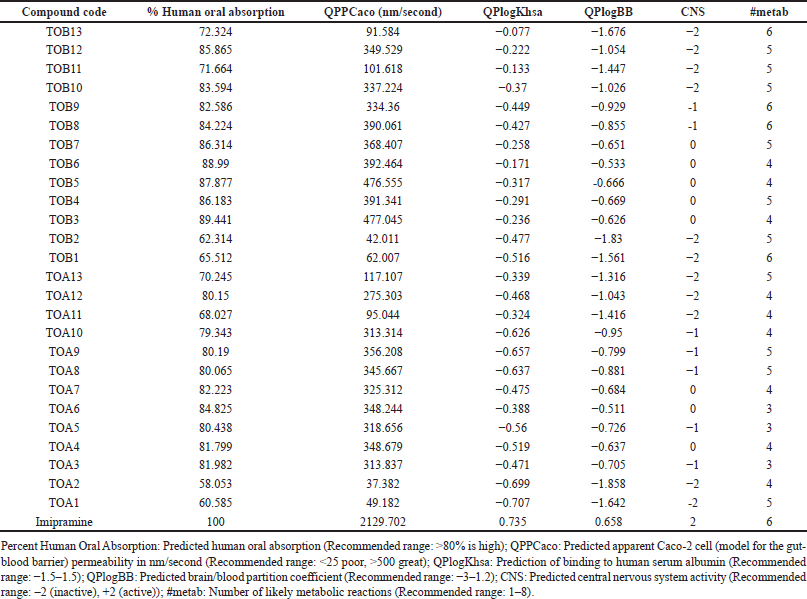 | Table 7. Predicted ADME profile. [Click here to view] |
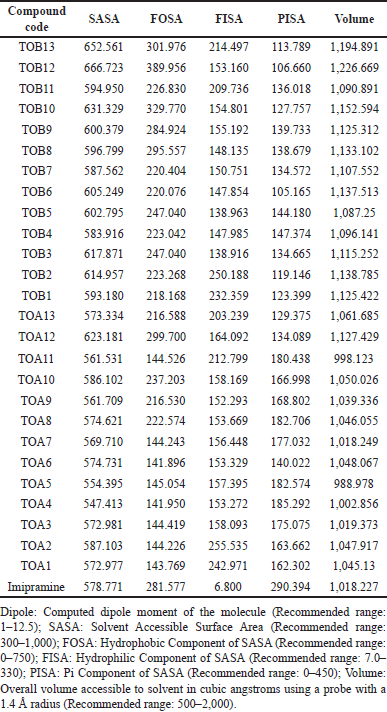 | Table 8. Predicted solvent-accessible surface area. [Click here to view] |
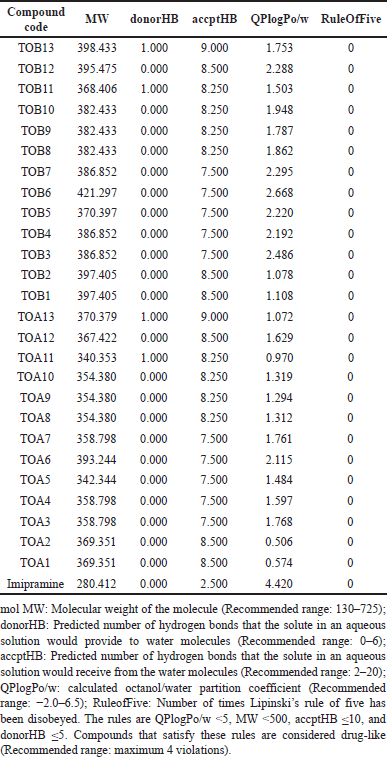 | Table 9. Validation of rule of five. [Click here to view] |
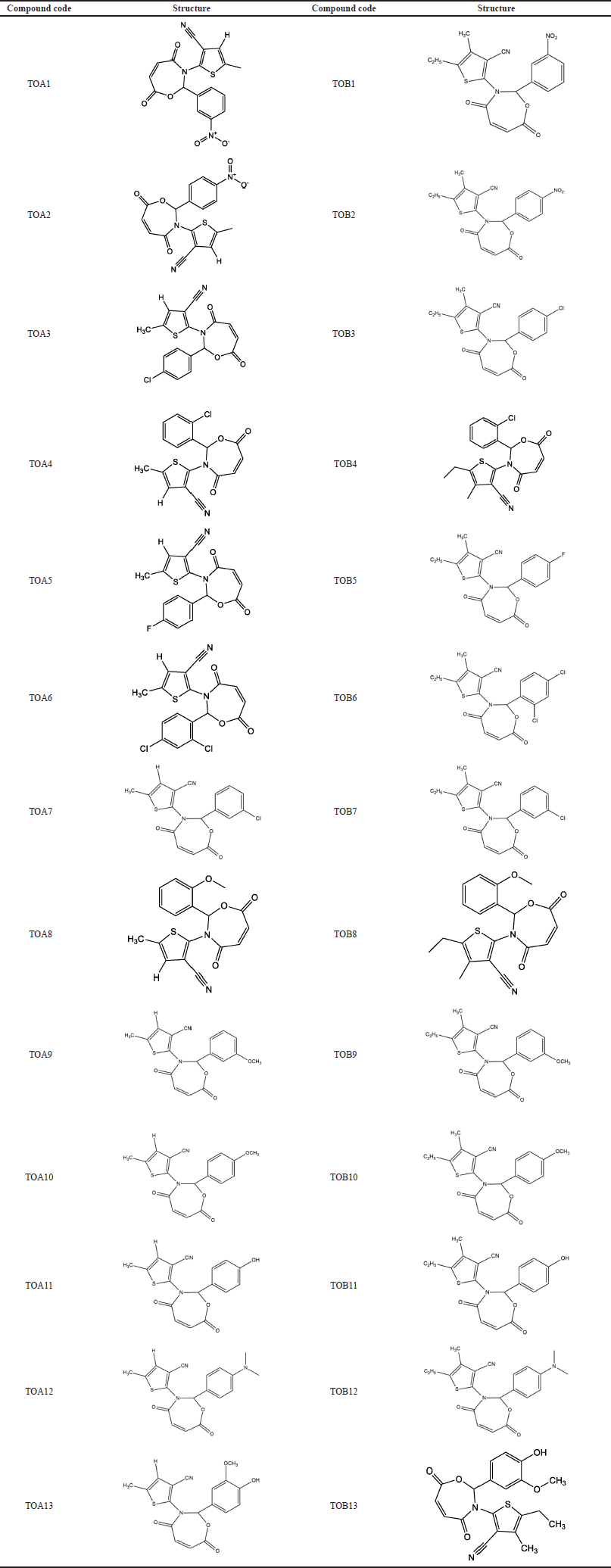 | Table 10. Compound code and structures of the molecules. [Click here to view] |
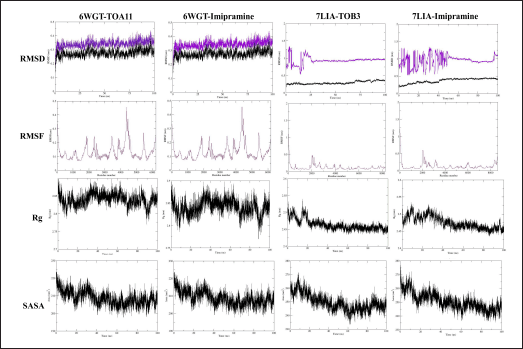 | Figure 6. MD simulation study. [Click here to view] |
Validation of rule of five
All the molecules were predicted to obey Lipinski’s rule of five which indicates good oral bioavailability of the compounds (Table 9).
MD simulation study
For 6WGT with TOA11 and imipramine the RMSD for the backbone and complex displayed fluctuation in the range of ~0.3 nm; and for 7LIA with TOB3 and imipramine showed the RMSD for the backbone and complex showed a fluctuation in the range of ~0.48 nm; similarly, the RMSF value fluctuated in the average of 0.82 nm throughout the simulation, which indicates the RMSF for c-alpha atoms is stable. The RoG for the complex was found to be on average for all complexes averaging 0.115 nm throughout the simulation. Additionally, SASA was found for 6WGT with TOA11 and imipramine and 7LIA with TOB3 and imipramine in the range of 198 to ~224, ~200 to ~223, 233 to ~272, ~236 to ~278 nm2, respectively (Fig. 6)
CONCLUSION
The present study was successful in designing molecules containing thiophene incorporated 1,3-oxazepine. The investigation involved 26 molecules which were analyzed in silico for the antidepressant activity when docked against the proteins 5-HT2AR (PDB ID: 6WGT) and 5-HTT (PDB ID: 7LIA). The compounds showed appreciable interactions like hydrophobic, polar, positive, negative, and hydrogen bonding with different amino acid residues of the proteins. The free energy of the ligand-receptor complexes was analyzed successfully. Pharmacophore modeling revealed the potential chemical features responsible for the interaction of potential molecules TOB3 and TOA11. The ADMET and physicochemical properties prediction revealed the drug-likeness of the molecules. MD simulation shows the stability and compactness of the complex between the designed ligands and targeted receptor and all the obtained values of RMSD, RMSF, SASA, and Rg were under acceptable ranges. Thus, the results of the study disclosed the scope for the synthesis, in-vivo evaluation, and development into the formulation of the designed compounds.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
ACKNOWLEDGMENT
The authors are thankful to Nitte (Deemed to be University) for providing the necessary facilities to carry out this research.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Bank RPD. RCSB PDB—7LIA: 5-HT bound serotonin transporter reconstituted in lipid nanodisc in presence of NaCl in outward facing conformation n.d. Available via https://www.rcsb.org/structure/7lia (Accessed 10 December 2022a)
Bank RPD. RCSB PDB—6WGT: crystal structure of HTR2A with hallucinogenic agonist n.d. Available via https://www.rcsb.org/structure/6WGT (Accessed 10 December 2022b)
Berrade L, Aisa B, Ramirez MJ, Galiano S, Guccione S, Moltzau LR, Levy FO, Nicoletti F, Battaglia G, Molinaro G, Aldana I, Monge A, Perez-Silanes S. Novel Benzo[ b] thiophene derivatives as new potential antidepressants with rapid onset of action. J Med Chem, 2011; 54:3086–90. doi: https://doi.org/10.1021/jm2000773
Brigitta B. Pathophysiology of depression and mechanisms of treatment. Dialogues Clin Neurosci, 2002; 4:7–20. doi: https://doi.org/10.31887/DCNS.2002.4.1/bbondy
Cascade E, Kalali AH, Kennedy SH. Real-world data on SSRI antidepressant side effects. Psychiatry, 2009; 6:16–8.
Depression. n.d. Available via https://www.who.int/news-room/fact-sheets/detail/depression (Accessed 24 November 2022)
Duffy EM, Jorgensen WL. Prediction of properties from simulations: free energies of solvation in hexadecane, octanol, and water. J Am Chem Soc, 2000; 122:2878–88. doi: https://doi.org/10.1021/ja993663t
Duval F, Lebowitz BD, Macher JP. Treatments in depression. Dialogues Clin Neurosci, 2006; 8:191–206. doi: https://doi.org/10.31887/DCNS.2006.8.2/fduval
Dwivedi PSR, Patil R, Khanal P, Gurav NS, Murade VD, Hase DP, Kalaskar MG, Ayyanar M, Chikhale RV, Gurav SS. Exploring the therapeutic mechanisms of Cassia glauca in diabetes mellitus through network pharmacology, molecular docking and molecular dynamics. RSC Adv, 2021; 11:39362–75. doi: https://doi.org/10.1039/D1RA07661B
Eison AS, Mullins UL. Regulation of central 5-HT2A receptors: a review of in vivo studies. Behav Brain Res, 1995; 73:177–81. doi: https://doi.org/10.1016/0166-4328(96)00092-7
Evans-Lacko S, Aguilar-Gaxiola S, Al-Hamzawi A, Alonso J, Benjet C, Bruffaerts R, Chiu WT, Florescu S, de Girolamo G, Gureje O, Haro JM, He Y, Hu C, Karam EG, Kawakami N, Lee S, Lund C, Kovess-Masfety V, Levinson D, Navarro-Mateu F, Pennell BE, Sampson NA, Scott KM, Tachimori H, Ten Have M, Viana MC, Williams DR, Wojtyniak BJ, Zarkov Z, Kessler RC, Chatterji S, Thornicroft G. Socio-economic variations in the mental health treatment gap for people with anxiety, mood, and substance use disorders: results from the WHO World Mental Health (WMH) surveys. Psychol Med, 2018; 48:1560–71. doi: https://doi.org/10.1017/S0033291717003336
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov, 2015; 10:449–61. doi: https://doi.org/10.1517/17460441.2015.1032936
Gibbs IS, Heald A, Jacobson H, Wadke D, Weliky I. Physical characterization and activity in vivo of polymorphic forms of 7-Chloro-5,11-dihydrodibenz[b,e],[1,4] oxazepine-5-carboxamide, a potential tricyclic antidepressant. J Pharm Sci, 1976; 65:1380–5. doi: https://doi.org/10.1002/jps.2600650929
Givens JL, Houston TK, Van Voorhees BW, Ford DE, Cooper LA. Ethnicity and preferences for depression treatment. Gen Hosp Psychiatry, 2007; 29:182–91. doi: https://doi.org/10.1016/j.genhosppsych.2006.11.002
Kalirajan R, Pandiselvi A, Gowramma B, Balachandran P. In-silico design, ADMET screening, MM-GBSA binding free energy of some novel isoxazole substituted 9-anilinoacridines as HER2 inhibitors targeting breast cancer. Curr Drug Res Rev, 2019; 11:118–28. doi: https://doi.org/10.2174/2589977511666190912154817
Kelly K, Posternak M, Jonathan EA. Toward achieving optimal response: understanding and managing antidepressant side effects. Dialogues Clin Neurosci, 2008; 10:409–18. doi: https://doi.org/10.31887/DCNS.2008.10.4/kkelly
Kendell RE. The classification of depressions: a review of contemporary confusion. Br J Psychiatry, 1976; 129:15–28. doi: https://doi.org/10.1192/bjp.129.1.15
Martin P, Waters N, Schmidt CJ, Carlsson A, Carlsson ML. Rodent data and general hypothesis: antipsychotic action exerted through 5-HT2A receptor antagonism is dependent on increased serotonergic tone. J Neural Transm, 1998; 105:365. doi: https://doi.org/10.1007/s007020050064
Mathew B, Suresh J, Anbazhagan S, Dev S. Proposed interaction of some novel antidepressant pyrazolines against monoamine oxidase isoforms. Molecular docking studies and PASS assisted in silico approach. Biomed Aging Pathol, 2014; 4:297–301. doi: https://doi.org/10.1016/j.biomag.2014.07.011
Mathew RJ, Weinman ML, Mirabi M. Physical symptoms of depression. Br J Psychiatry, 1981; 139:293–6. doi: https://doi.org/10.1192/bjp.139.4.293
Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem, 1994; 40:288–95. doi: https://doi.org/10.1093/clinchem/40.2.288
Paykel ES. Basic concepts of depression. Dialogues Clin Neurosci, 2008; 10:279–89. doi: https://doi.org/10.31887/DCNS.2008.10.3/espaykel
Schrödinger | Schrödinger is the scientific leader in developing state-of-the-art chemical simulation software for use in pharmaceutical, biotechnology, and materials research. n.d. Available via https://www.schrodinger.com/ (Accessed 10 December 2022).
Shah R, Verma PK. Therapeutic importance of synthetic thiophene. Chem Cent J, 2018; 12:137. doi: https://doi.org/10.1186/s13065-018-0511-5
Sharma G, Park JY, Park MS. Synthesis and anticonvulsant evaluation of 6-amino-1,4-oxazepane-3,5-dione derivatives. Arch Pharm Res, 2008; 31:838–42. doi: https://doi.org/10.1007/s12272-001-1235-0
Squire LR, editor. Fundamental neuroscience. 3rd ed, Elsevier/Academic Press Amsterdam, The Netherlands/Boston, MA, 2008.
Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev, 2019; 119:9478–508. doi: https://doi.org/10.1021/acs.chemrev.9b00055
Wardakhan W, Abdel-Salam O, Elmegeed G. Screening for antidepressant, sedative and analgesic activities of novel fused thiophene derivatives. Acta Pharm, 2008; 58:1–14. doi: https://doi.org/10.2478/v10007-007-0041-5