
INTRODUCTION
In late December 2019, a pneumonia outbreak of unidentified etiology was announced in Wuhan, Hubei Province, People’s Republic of China. The most common symptoms reported in patients infected with such disease are fever, cough, fatigue, myalgia, diarrhea, and shortness of breath; some cases lead to acute and fatal respiratory syndromes (Ciotti et al., 2019; Hamed, 2020). World Health Organization (WHO) later named it coronavirus disease 2019 (COVID-19), which was caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Zhang et al., 2020). The number of people infected with the virus has increased rapidly around the globe (Ouassou et al., 2020). As of December 6, 2022, 641,915,931 confirmed cases of COVID-19, including 6,622,760 deaths, were reported to WHO (2022). In addition, COVID-19 has also caused socioeconomic impacts, including physical distancing, self-quarantine, and travel restrictions that resulted in a considerable slowdown in economic activities (Nicola et al., 2020).
The development of rapid diagnostic tests, vaccines, and antiviral drugs is essential to detect, prevent, and treat COVID-19 (Hamed, 2020). In terms of treatment, various viral proteases which are essential for viral replication have been extensively studied as drug targets, including the main protease or 3-chymotrypsin-like protease (Mpro/3CLpro), papainlike protease (PLpro), angiotensin-converting enzyme 2 (ACE-2), RNAdependent RNA polymerase (RdRp), spike protein, serine protease, and helicase (Illian et al., 2021). After SARS-CoV-2 infection, two viral polyproteins (pp1a and pp1ab), containing 11 or 16 nonstructural proteins (NSPs), are produced. Mpro, also known as nsp5, plays a critical role in cleaving eleven sites along these polyproteins. It represents crucial processing actions for viral construction and maturation (Hardianto et al., 2021; Lee et al., 2020; Suárez and Díaz, 2020). Therefore, inhibiting SARS-CoV-2 Mpro is needed to block the viral assembly and replication (Das et al., 2021).
Azadirachta indica A. Juss, commonly known as Neem, is a member of the Meliaceae family, which originated from South and Southeast Asia, and is now grown in other tropical and subtropical regions, including Africa, America, and Australia (Passos et al., 2019). It is one of the most versatile medicinal plants having a wide range of pharmacological activities, such as antioxidant, anti-inflammatory, antidiabetic, anticancer, antimalarial, antifungal, antibacterial, antiviral, hepatoprotective, neuroprotective, and wound healing effect activities (Alzohairy, 2016). Azadirachtin, nimbolide, and gedunine are some of the bioactive compounds of A. indica known to have a stupendous ability to arrange many biological pathways in vitro and in vivo (Sarkar et al., 2021). Several reports were showing the antiviral properties of A. indica against coxsackievirus B (Badam et al., 1999), herpes simplex virus type 1 (Tiwari et al., 2010), human immunodeficiency virus type 1 (Awah et al., 2011), dengue virus type 2 (Dwivedi et al., 2021), and poliovirus type 1 (Baildya et al., 2021; Faccin-Galhardi et al., 2012).
Computer-aided drug discovery approaches are a cost-effective and time-saving strategy to screen candidates with inhibitory potency targeting SARS-CoV-2. Furthermore, molecular docking and virtual screening techniques have been demonstrated to identify potential drugs for COVID-19 treatment by the protein-ligand free energy of binding estimation (Hosseini et al., 2021). Sharon (2020) has reported the potency of selected compounds from A. indica in inhibiting SARS-CoV-2 Mpro (PDB code: 6Y2E, 6LU7, and 2GTB) using molecular docking. Among 13 bioactive molecules, azadiradione, epiazadiradione, nimbione, and vepnin seemed to have the highest docking score (Sharon, 2020). Another study also showed that azadirachtin, epoxyazadiradione, and gedunin are the compounds that give the highest docking score (Kumar, 2020).
The molecular docking technique has restrictions on protein flexibility, solvation models, and its simplification of the scoring functions (Souza et al., 2021). Thus, another computational method is vital to complement molecular docking to give more reliable results. Full atomistic molecular dynamics simulation (MDS) is one of the methods for analyzing the dynamic behavior of ligand, protein, and solvent at a fixed period (Santos et al., 2019).
In the current work, a more extensive data set of secondary metabolites than in former studies (Kumar, 2020; Sharon, 2020) was utilized to explore the potency of A. indica against SARS-CoV-2 Mpro (PDB code: 6M0K) through molecular docking and MDSs. Rather than using an aspartate protease inhibitor as positive control like in those studies, we compared the testing ligands using a specific 3CL-like protease inhibitor, nirmatrelvir (NTV), for positive control in the docking and MDS. The 3CLpro is the Mpro of SARS-COV-2. It has the characteristic of the catalytic dyad Cys-His, which differs from the catalytic triad Asp-Thr-Gly of aspartate protease. Thus, using NTV as positive control is more reasonable.
MATERIALS AND METHODS
3D structures preparation and geometry optimization
Names of secondary metabolites contained in different parts of A. indica were searched from the references. The structures were obtained from the simplified molecular-input line-entry system (SMILES) in PubChem (https://pubchem.ncbi.nlm.nih.gov/) and converted into 2D structures in ChemDraw® Professional 20.1.1.125 (https://perkinelmerinformatics.com/). Some of the structures, not found in PubChem, were drawn in ChemDraw® Professional 20.1.1.125 manually, referring to the literature. Afterward, the conversion of 3D from 2D structures was executed using Chem3D® Ultra 20.1.1.125 (https://perkinelmerinformatics.com/). The 3D structure of NTV, an effective and safe antiviral drug that inhibits the SARS-CoV-2 Mpro (Hung et al., 2022), was also prepared in a similar procedure. The geometry of 3D structures was, later optimized by a semiempirical method using the Parametric Method 7 (PM7) in MOPAC2016™ (http://openmopac.net/) (Stewart, 2016).
The 3D structure of SARS-CoV-2 Mpro (PDB code: 6M0K) (Dai et al., 2020) was downloaded from RCSB protein data bank (https://www.rcsb.org/) (Berman et al., 2000) and prepared by AutoDockTools 1.5.7 (https://ccsb.scripps.edu/mgltools/) (Sanner, 1999). The water molecules were discarded, and the reference ligand, FJC, was separated from 6M0K. The target protein was added with polar hydrogens, merged with nonpolar hydrogens, added Gasteiger charges, and saved in PDBQT format. Additionally, the reference ligand was converted to PDBQT format as well.
Molecular docking
The molecular docking was conducted in AutoDock 4.2 (https://autodock.scripps.edu/) (Morris et al., 2009) on a high-performance computer with Debian GNU/Linux 8.11 (jessie) 64-bit operating system, Intel® Core™ i7-5820K CPU @ 3.30 GHz × 12 processors, 31.4 GiB memory, and NVIDIA® GeForce GTX™ 980 Ti/PCIe/SSE2 graphics card. First, the redocking of the reference ligand, FJC, on the target protein was necessary for molecular docking method validation. Subsequently, all prepared secondary metabolites and NTV were docked to the target protein in the same parameters as molecular docking validation. The molecular docking parameters used were central grid point coordinates x = −12.045, y = 11.235, z = 68.856; grid box size 40 × 46 × 40; grid point spacing 0.375 Å; default docking parameters; and Lamarckian Genetic Algorithm (Morris et al., 1998). Nonbonded interactions between ligand and SARS-CoV-2 Mpro for the molecular docking result structures with the lowest free energy of binding, NTV, and FJC were analyzed with BIOVIA Discovery Studio 2021 Visualizer (https://www.3ds.com/).
Molecular dynamics simulation
The MDS were performed using GROMACS 2021.5 (https://www.gromacs.org/) (Berendsen et al., 1995) on Dell Precision 5820 Tower X-Series with Ubuntu 20.04 (Focal Fossa) 64-bit operating system, Intel® Core™ i9-10900X CPU @ 4.60 GHz × 20 processors, 62.5 GiB memory, and NVIDIA® Quadro RTX™ 8000 graphics card. The protein topology was written by pdb2gmx using CHARMM36-feb2021 (http://mackerell.umaryland.edu/) (Huang et al., 2017) force field and TIP3P original water model (Jorgensen et al., 1983). The topologies of the minimum energy configurations of the three best secondary metabolites in A. indica reference ligand, and NTV obtained from molecular docking studies were processed in CHARMM General Force Field server (https://cgenff.umaryland.edu/) (Vanommeslaeghe et al., 2010; Yu et al., 2012). Furthermore, the topology of the Mpro-ligand complex was built by combining Mpro and ligand topologies.
The unit cell was defined using a rhombic dodecahedral shape, and the Mpro-ligand complex was placed at its center at least 10 Å from the unit cell shape edge. Besides, this unit cell was filled with water using spc216.gro, a generic equilibrated three-point solvent configuration for TIP3P water. Using genion module in GROMACS 2021.5, some water molecules were replaced with Na+ and Cl− to make the system charge neutral. Hereafter, energy minimization was accomplished by the steepest descent algorithm until the maximum force was under 1,000 kJ mol−1 nm−1. Two equilibration phases were carried out, isothermal-isochoric ensemble (NVT) and isothermal-isobaric ensemble (NPT). Both NVT and NPT equilibration phases were done for 100 ps at 300 K. Fast smooth particle-mesh Ewald (PME) (Darden et al., 1993) was applied for long-range electrostatic. In contrast, the cut-off of short-range electrostatic and van der Waals was determined at 12 Å. After the system was well equilibrated at 300 K and 1 bar, the 100-ns molecular dynamics production was run in the same parameters as NPT. However, the position restraints were released previously. The temperature and pressure coupling used for the production were modified Berendsen thermostat (v-rescale) (Bussi et al., 2007) and the Parrinello-Rahman barostat (Parrinello and Rahman, 1981). Unrestrained MDS runs were conducted in triplicate for each ligand. The MDS was supported with the graphical processing unit, where the PME, nonbonded force calculation, and bonded force calculation could be offloaded, which accelerated the simulation significantly.
MDS trajectory analysis and binding energy calculation
The MDS structural trajectories were recentered and rewrapped within the rhombic dodecahedral shape using the gmx trjconv module. gmx rms module was computed for the root mean square deviation (RMSD) to compare each structure from a trajectory to the energy minimization result structure. In addition, the root mean square fluctuation (RMSF) of the residue position in the trajectory after fitting to the energy minimization result frame was analyzed with the gmx rmsf module.
The binding free energy values of protein-ligand complexes were estimated by molecular mechanics generalized born surface area (MMGBSA) method using gmx_MMPBSA (https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/) (Valdés-Tresanco et al., 2021). The MMGBSA binding free energy (ΔGoMMGBSA) in solution is defined as follows in
where each Gx term on the right in the equation is calculated as follows in
EMM is the molecular mechanical energy in the gas phase and Gsolvation is the solvation energy. A single trajectory protocol approach was utilized for MMGBSA calculation, where the receptor and the ligand trajectories were extracted from the complex. Consequently, the internal energy terms, including angles, bonds, and dihedrals, were excluded as these are similar in bound and unbound states (Valdés-Tresanco et al., 2021).
Drug-likeness and absorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction
The drug-likeness of secondary metabolites in A. indica was evaluated using Lipinski’s rule of five in ChemOffice 2020 (https://perkinelmerinformatics.com/). The rule applies five criteria to specify if a compound is drug-like or not. The compound must have a molecular weight lower than 500 Da, log P under 5, h-bond donors fewer than or equal to 5, h-bond acceptors fewer than or equal to 10, and the violation from mentioned four rules not more than one. Eventually, pharmacokinetic properties, including ADMET, were predicted using the pkCSM server (http://biosig.unimelb.edu.au/pkcsm/prediction) (Pires et al., 2015).
RESULTS AND DISCUSSION
Molecular docking
A total of 76 secondary metabolites in different parts of A. indica were gathered from the literature (Table S1) (Akhila and Rani, 1999; Atawodi and Atawodi, 2009; Chan et al., 1973; Girish and Shankara, 2008; Govindachari et al., 2000, 1996; Govindachari and Geetha, 1997; Herrera-Calderon et al., 2019; Kumar et al., 2014; Luo et al., 2000; Siddiqui et al., 2003, 1992; Singh and Sharma, 2020; Singh, 2009). The geometry of these compounds was optimized to reach the lowest energy and subsequently form the most stable poses (Tripathy and Sahu, 2018). In this study, PM7 was applied as the semiempirical geometry optimization method due to its high docking positioning accuracy (Sulimov et al., 2022, 2019). The geometry optimization results of NTV and three secondary metabolites with the highest molecular docking score are shown in Figure S1.
Molecular docking studies were performed on all secondary metabolites, NTV, and FJC. In advance, a validation step was conducted by redocking FJC as the reference ligand on SARS-CoV-2 Mpro. This is intended to verify whether the docking algorithm results in an accurate pose and whether the scoring function recognizes it as a top pose (Prieto-Martínez et al., 2018). It was confirmed to be valid if the RMSD of crystal and redocked structures is less than 2 Å (Tallei et al., 2020). The best RMSD achieved from this step was 1.938 Å. Crystal and redocked structures of reference ligand in the overlay are depicted in Figure S2.
Virtual screening based on a molecular docking approach was employed to identify potential compounds contained in A. indica. In molecular docking simulation, the energy of ligand-protein complex structure must be the global minimum on the binding energy landscape, representing a thermodynamic state of the energy function used in the simulation (Verkhivker et al., 2000). The molecular docking studies showed that among 76 secondary metabolites, 18 of them have higher binding energy scores than NTV. NTV was used in this study as this antiviral component of PAXLOVID™ which has SARS-CoV-2 Mpro enzyme inhibitory activity (Catlin et al., 2022). Odoratone (ORN), salimuzzalin (SMZ), and nimbocidin2 (NC2) had the highest binding energy scores of −11.57, −9.83, and −9.60 kcal/mol, respectively. In comparison, binding energy scores for NTV and FJC are −8.42 and −7.93 kcal/mol, respectively. The estimated free energy of binding for all compounds is shown in Table S2, while the top 15 rankings, reference ligand, and NTV are listed in Table 1.
The two best secondary metabolites from molecular docking studies, ORN, and SMZ, were selected for visualization. Figure 1a shows that ORN built conventional hydrogen bonds with several residues, i.e., His163, Gln189, Thr190, and Gln192. Hydrogen bonding is principal intermolecular interactions that provide plenty of stability to the protein-ligand complex (Kwofie et al., 2021). Other critical amino acids involved in hydrophobic interactions (alkyl bonds) are Met165 and Pro168. Hydrophobic interaction plays an essential role as a driving force that allows the spontaneous folding of the protein into 3D structures (Yusof et al., 2019).
NTV and FJC from molecular docking results were also visualized in Figure 1c and d, respectively. In contrast to ORN and SMZ, Gln192 was linked via an unfavorable donor-donor bond with NTV. Besides, NTV and FJC have halogen (fluorine) bonds that ORN and SMZ do not have. The halogen bonding has stabilization effects on inter- and intramolecular interactions and influence molecular folding (Suárez-Castro et al., 2018). These bonding types were found in Thr26 connected to NTV as well as Arg188 and Gln189 linked to FJC.
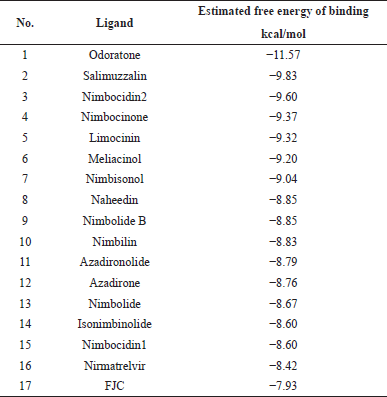 | Table 1. Estimated free energy of binding rankings from molecular docking results of secondary metabolites in A. indica (top 15 of 76 compounds), reference ligand, and NTV. [Click here to view] |
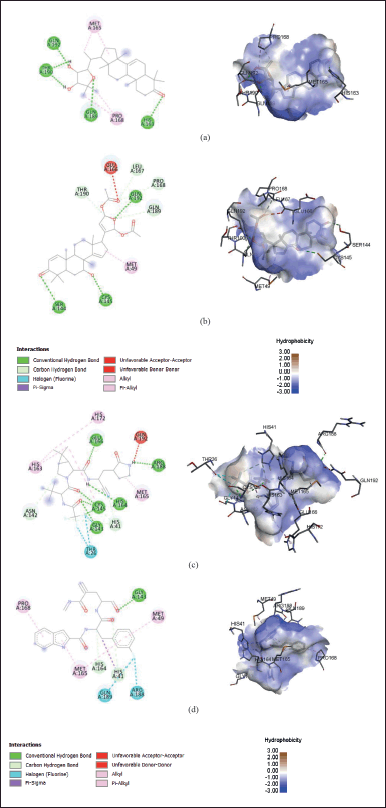 | Figure 1. Protein-ligand interactions connecting residues from SARS-CoV-2 Mpro (6M0K) and (a) ORN, (b) SMZ, (c) NTV, and (d) the reference ligand (FJC) in 2D and 3D structures from molecular docking results. [Click here to view] |
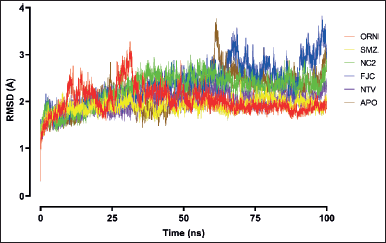 | Figure 2. RMSD plots of SARS-CoV-2 Mpro backbone in ligand-unbound (APO) and ligand-bound structures. The given chart is the average of the triplicate MDS results for each ligand. [Click here to view] |
Molecular dynamics simulation
The stability of complexes in the SARS-CoV-2 Mpro active site was studied using MDS within 100 ns. The ligands to be compared in this simulation are ORN, SMZ, NC2, reference ligand (FJC), and NTV as a positive control. Parameters, such as RMSD, RMSF, and binding energy calculation (MMGBSA), generated from MDS were analyzed.
The RMSD value measures the simulation system stability and the conformational perturbation of the protein backbone during a time scale of simulation (Sargsyan et al., 2017). Overall, the convergence of RMSD for the complex of SARS-CoV-2 Mpro and the ligands were observed after 15 ns with the average RMSD value of 2.10 Å that denotes fewer changes in the overall structure during the simulation time (Fig. 2). The protein backbone of SARS-CoV-2 Mpro was stable during the entire simulation, with the average RMSD value of 2.16 Å, which indicates lower fluctuation and stable behavior. The ligand SMZ displayed the most stable simulation system with the lowest average RMSD value of 1.90 Å among other ligands, slightly lower than positive control NTV (1.98 Å) for the SARS-CoV-2 Mpro backbone perturbation. The RMSD profile of the complex Mpro-SMZ resembles that of the complex Mpro-NTV. The difference between the two is that no residues in the backbone of Mpro-SMZ appeared to have RMSD > 2.50 Å, while some residues in the backbone of Mpro-NTV were still perturbed higher than 2.50 Å. The docked complex of the other ligand, ORN, was stabilized without significant deviation since 50 ns with the average RMSD value of 2.02 Å. Unlike other complexes that have been quite stable since the simulation’s beginning, the complex with ligand ORN resulted in an RMSD of more than 3.00 Å before reaching 50 ns. On the other hand, the complex Mpro-FJC, the reference ligand, showed significant deviation with RMSD > 3.00 Å from 58 ns onward and average backbone RMSD 2.36 Å, putting this complex as the most unstable among other ligands. NC2, the third-best secondary metabolite in the docking study, still remained in third place in this MDS compared to SMZ and ORN with the average RMSD 2.26 Å, but still better than the reference ligand, FJC.
Atomic fluctuation of each residue was evaluated through RMSF to understand the residue behavior of SARS-CoV-2 Mpro in complex with the ligands. The higher the peak, the higher the atomic fluctuation (Fig. 3). The atoms in N- and C-terminal have higher fluctuation, up to 8 Å, than the atoms in any other area of the protein. The atoms in the protein surface loops tend to fluctuate more than the atoms in the α-helix or β-strand, especially those in the buried area, which indicates a more flexible movement of the protein loops. Some residue types, such as arginine, phenylalanine, glutamine, and tyrosine, located in the surface loops have RMSF values between 2 and 4 Å compared to other residue types in the same location that have lower RMSF values. This could be due to their longer side chain that interacts with polar water molecules.
The atomic fluctuation in the ligand binding site was also evaluated to know ligand binding stability inside the pocket. It was revealed that SMZ generated the lowest average RMSF value of 1.32 Å when interacting with residues in the ligand binding site, followed by NTV (1.42 Å), ORN (1.43 Å), NC2 (1.62), and reference ligand, FJC, (1.71 Å). The average RSMF value of residues in the ligand binding site for SMZ, NTV, and ORN is even lower than that of the Apolipoprotein-SARS-CoV-2 Mpro (1.61 Å). Residues in the ligand binding site with high RSMF values are predicted to make weaker interactions with the ligand. It implies that SMZ produces the most optimal binding when interacting with SARS-CoV-2 Mpro. Some residues in the ligand binding site whose atoms fluctuate more than 2 Å when interacting with SMZ are only Met49, Arg188, and Gln189, of which the last two residues happen to be in the surface loop. The other residues in the ligand binding site whose atoms have RMSF lower than 2 Å might provide stronger binding. Interestingly, ORN might provide stronger binding because all residues in its binding site have atomic fluctuation below 2 Å, although the average value is not better than SMZ.
The ΔGoMMGBSA calculation was carried out to 100 ns of MDS trajectory (0–10,001 frames). Figure 4 shows that ORN had the most significant average ΔGoMMGBSA value, −31.27 kcal/mol, compared to other ligands. Meanwhile, the reference ligand, FJC, had a more negative average ΔGoMMGBSA value than two other compounds from A. indica, SMZ and NC2, and the positive control, NTV. The calculated average ΔGoMMGBSA values of FJC, SMZ, NC2, and NTV were found to be −26.46, −20.73, −16.30, and −17.32 kcal/mol, respectively. The calculated values are approximations of the binding free energy, where more negative values indicate stronger binding.
Drug-likeness and ADMET prediction
Similar to the efficacy of plants, bioavailabilities and ADMET profiles are also determined by the nature of each compound contained. It is essential to evaluate these properties so a single compound can be suggested as a drug candidate that is safe and optimal for use. However, ADME studies are not usually required for herbal remedy discovery and development. Most herbal medicines have little or no data on their ADME and pharmacokinetic properties in humans (He et al., 2011).
Lipinski’s rule or rule-of-five states that poor absorption or permeability of a compound is more likely when it has more than 5 hydrogen-bond donors, the molecular mass is higher than 500, the calculated log P is greater than 5, and the hydrogen bond acceptor is more than 10. Lipinski’s rule of five was initially conceived to aid the development of orally administrated drugs. However, it was not intended to generalize all small-molecule drugs (Lipinski et al., 1997; Neidle, 2012). From the evaluation of 15 metabolites with the highest rankings as potential inhibitors of SARS-CoV-2 Mpro, only limocinin did not meet this rule. Lipinski’s rule-of-five results for all secondary metabolites are listed in Table S3.
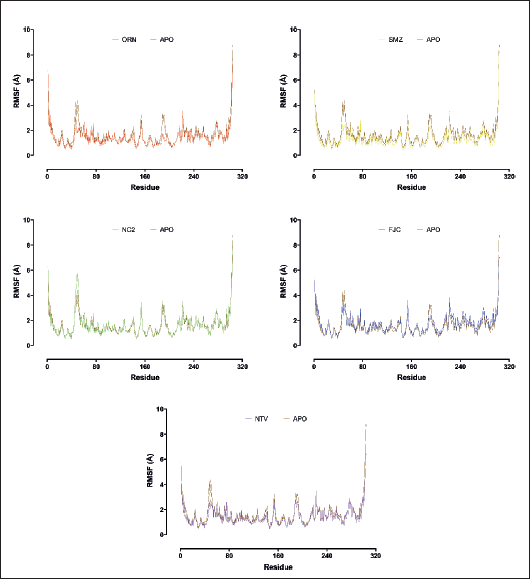 | Figure 3. RMSF plots of SARS-CoV-2 Mpro in ligand-unbound (APO) and ligand-bound structures. The given charts are the average of the triplicate MDS results for each ligand. [Click here to view] |
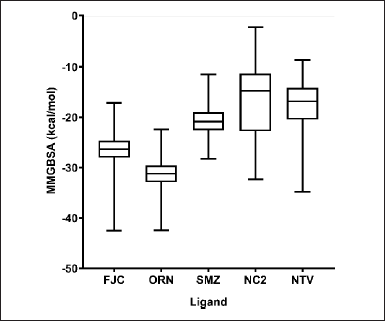 | Figure 4. ΔGoMMGBSA values’ box plot of some ligands binding to SARS-CoV-2 Mpro, which are the averages of the triplicate MDS results for each ligand. [Click here to view] |
The predicted ADMET properties are tabulated in Tables S4–S7. The prediction data shows that each compound from A. indica displayed diverse ADMET. Still, it can be tolerated because there are synergy effects probabilities. In general, they tend to be absorbed by the gastrointestinal part sufficiently with low blood–brain–barrier (BBB) ??permeability values. Some compounds do not affect CYP2D6 substrate, CYP2C9 inhibitor CYP2D6 inhibitor cytochromes. The skin permeability for the compounds is between 2,314 and 3,599 units. Most compounds show negative AMES toxicity except nimbisonol, and none of them has shown hERGI inhibition activity. The LD50 values of the studied compounds are between 1.8 and 4.0 mol/kg. The chronic oral rat toxicity in the lowest observed adverse effect Level (LOAEL) values lie between 0.1 and 3.5 (log mg/kg bw/day). Most secondary metabolites in A. indica do not show any hepatotoxicity except nimbic acid, nimbic acid B, nimbidic acid, nimbilin, nimbinic acid, nimbinene, and margolone. The compound of 2,6-bis-(1,1)-dimethyl ethyl-4-methyl phenol showed skin sensitization.
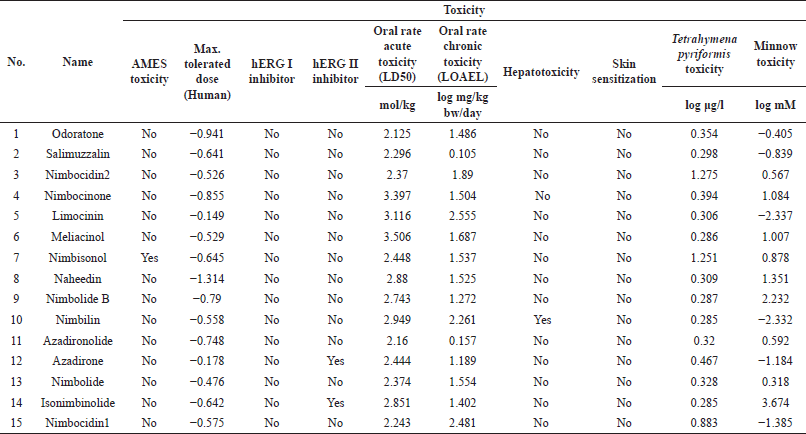 | Table 2. Toxicity prediction for secondary metabolites in A. indica (top 15 of 76 compounds from molecular docking results). [Click here to view] |
ORN, as the most potent candidate, shows one violation of Lipinski’s rule due to its log P (more than 5). This should attract attention because it frequently leads to compounds with rapid metabolic turnover, low solubility, and poor absorption. It is supported by the low value of Caco-2 permeable (in vitro model of the human intestinal mucosa). The logarithm of the apparent permeability coefficient (log Papp; log cm/s) is lower than 0.9. Fortunately, the predictions of intestinal absorption and skin permeability are considerably high.
Moreover, ORN shows a low BBB permeability value. It does not affect CYP2D6 substrate, CYP1A2 inhibitor, CYP2C9 inhibitor, or CYP2D6 inhibitor cytochromes. This compound is predicted to have adverse AMES toxicity and hERG inhibitor. It also may not affect hepatotoxicity and skin sensitization. The toxicity prediction of the 15 metabolites with the highest docking score is summarized in Table 2.
CONCLUSION
Virtual screening on 76 secondary metabolites in A. indica to identify their potency as SARS-CoV-2 Mpro inhibitors were done through molecular docking and MDS. In molecular docking studies, ORN, SMZ, and NC2 were known to have the best binding energy scores and even better than NTV, an orally administrated SARS-CoV-2 Mpro inhibitor, and the reference ligand. Meanwhile, SMZ showed fascinating performance on MDS with the lowest average RMSD for the SARS-CoV-2 Mpro backbone perturbation. The ligand SMZ also had the lowest average RMSF, indicating its strong interactions with the residues. Nevertheless, in MDS studies, ORN showed the best average ΔGoMMGBSA value, which indicated the strongest binding free energy values of the protein-ligand complexes. This study showed that A. indica has several prospective compounds as SARS-CoV-2 Mpro inhibitors, and further wet experiments are needed to validate these potentials.
ACKNOWLEDGMENTS
This work was supported by the Budget Execution (Allotment) Document (DIPA) program of the Agency for the Assessment and Application of Technology (BPPT) 2021.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Akhila A, Rani K. Chemistry of the neem tree (Azadirachta indica A. Juss.). Fortschr Chem Org Naturst, 1999; 78:47–149. CrossRef
Alzohairy MA. Therapeutics role of Azadirachta indica (Neem) and their active constituents in diseases prevention and treatment. Evid Based Complement Altern Med, 2016; 2016:7382506. CrossRef
Atawodi SE, Atawodi JC. Azadirachta indica (neem): a plant of multiple biological and pharmacological activities. Phytochem Rev, 2009; 8:601–20. CrossRef
Awah FM, Uzoegwu PN, Ifeonu P. In vitro anti-HIV and immunomodulatory potentials of Azadirachta indica (Meliaceae) leaf extract. Afr J Pharm Pharmacol, 2011; 5:1353–9. CrossRef
Badam L, Joshi SP, Bedekar SS. “In vitro” antiviral activity of neem (Azadirachta indica. A. Juss) leaf extract against group B coxsackieviruses. J Commun Dis, 1999; 31:79–90.
Baildya N, Khan AA, Ghosh NN, Dutta T, Chattopadhyay AP. Screening of potential drug from Azadirachta indica (Neem) extracts for SARS-CoV-2: an insight from molecular docking and MD-simulation studies. J Mol Struct, 2021; 1227:129390. CrossRef
Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun, 1995; 91:43–56. CrossRef
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res, 2000; 28:235–42. CrossRef
Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys, 2007; 126:14101. CrossRef
Catlin NR, Bowman CJ, Campion SN, Cheung JR, Nowland WS, Sathish JG, Stethem CM, Updyke L, Cappon GD. Reproductive and developmental safety of nirmatrelvir (PF-07321332), an oral SARS-CoV-2 Mpro inhibitor in animal models. Reprod Toxicol, 2022; 108:56–61. CrossRef
Chan WR, Gibbs JA, Taylor DR. Triterpenoids from Trichilia havanensis Jacq. part I. The acetates of havanensin and trichilenone, new tetracarbocyclic tetranortriterpenes. J Chem Soc Perkin Trans 1, 1973:1047–50. CrossRef
Ciotti M, Angeletti S, Minieri M, Giovannetti M, Benvenuto D, Pascarella S, Sagnelli C, Bianchi M, Bernardini S, Ciccozzi M. COVID-19 outbreak: an overview. Chemotherapy, 2019; 64:215–23. CrossRef
Dai W, Zhang B, Jiang X-M, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C, Li Y, Bai F, Wang H, Cheng X, Cen X, Hu S, Yang X, Wang J, Liu X, Xiao G, Jiang H, Rao Z, Zhang LK, Xu Y, Yang H, Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science, 2020; 368:1331–5. CrossRef
Darden T, York D, Pedersen L. Particle mesh Ewald: an N? log (N) method for Ewald sums in large systems. J Chem Phys, 1993; 98:10089–92. CrossRef
Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn, 2021; 39:3347–57. CrossRef
Dwivedi VD, Bharadwaj S, Afroz S, Khan N, Ansari MA, Yadava U, Tripathi RC, Tripathi IP, Mishra SK, Kang SG. Anti-dengue infectivity evaluation of bioflavonoid from Azadirachta indica by dengue virus serine protease inhibition. J Biomol Struct Dyn, 2021; 39:1417–30. CrossRef
Faccin-Galhardi LC, Yamamoto KA, Ray S, Ray B, Linhares REC, Nozawa C. The in vitro antiviral property of Azadirachta indica polysaccharides for poliovirus. J Ethnopharmacol, 2012; 142:86–90. CrossRef
Girish K, Shankara BS. Neem–a green treasure. Electron J Biol, 2008; 4:102–11.
Govindachari TR, Geetha G. 13,14-desepoxyazadirachtin-A, a tetranortriterpenoid from Azadirachta indica. Phytochemistry, 1997; 45:397–9. CrossRef
Govindachari TR, Gopalakrishnan G, Suresh G. Isolation of various azadirachtins from neem oil by preparative high performance liquid chromatography. J Liq Chromatogr Relat Technol, 1996; 19:1729–33. CrossRef
Govindachari TR, Suresh G, Gopalakrishnan G. Insect antifeedant and growth regulating activities of neem seed oil—the role of major tetranortriterpenoids. J Appl Entomol, 2000; 124:287–91. CrossRef
Hamed MA. An overview on COVID-19: reality and expectation. Bull Natl Res Cent, 2020; 44:1–10. CrossRef
Hardianto A, Yusuf M, Hidayat IW, Ishmayana S, Soedjanaatmadja UMS. Exploring the potency of Nigella sativa seed in inhibiting SARS-CoV-2 main protease using molecular docking and molecular dynamics simulations. Indones J Chem, 2021; 21:1252–62. CrossRef
He S-M, Chan E, Zhou S-F. ADME properties of herbal medicines in humans: evidence, challenges and strategies. Curr Pharm Des, 2011; 17:357–407. CrossRef
Herrera-Calderon O, Ejaz K, Wajid M, Shehzad M, Tinco-Jayo JA, Enciso-Roca E, Franco-Quino C, Yuli-Posadas RÁ, Chumpitaz-Cerrate V. Azadirachta indica: antibacterial activity of neem against different strains of bacteria and their active constituents as preventive in various diseases. Pharmacogn J, 2019; 11:1597–604. CrossRef
Hosseini M, Chen W, Xiao D, Wang C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis Clin Med, 2021; 4:1–16. CrossRef
Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL, Grubmüller H, MacKerell AD Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods, 2017; 14:71–3. CrossRef
Hung Y-P, Lee J-C, Chiu C-W, Lee C-C, Tsai P-J, Hsu I-L, Ko WC. Oral nirmatrelvir/ritonavir therapy for COVID-19: the dawn in the dark? Antibiotics, 2022; 11:220. CrossRef
Illian DN, Siregar ES, Sumaiyah S, Utomo AR, Nuryawan A, Basyuni M. Potential compounds from several Indonesian plants to prevent SARS-CoV-2 infection: a mini-review of SARS-CoV-2 therapeutic targets. Heliyon, 2021; 7:e06001. CrossRef
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys, 1983; 79:926–35. CrossRef
Kumar AHS. Molecular docking of natural compounds from tulsi (Ocimum sanctum) and neem (Azadirachta indica) against SARS-CoV-2 protein targets. Biol Eng Med Sci Rep, 2020; 6:11–3. CrossRef
Kumar ASS, Bose KSC, Kumar KP, Raghavan S, Murali PM. Terpenoids and its commercial utility from Neem: the nature’s own pharmacy. Asian J Chem, 2014; 26:4940. CrossRef
Kwofie SK, Dolling NNO, Donkoh E, Laryea GM, Mosi L, Miller WA, Adinortey MB, Wilson MD. Pharmacophore-guided identification of natural products as potential inhibitors of Mycobacterium ulcerans cystathionine γ-synthase MetB. Computation, 2021; 9:32. CrossRef
Lee J, Worrall LJ, Vuckovic M, Rosell FI, Gentile F, Ton A-T, Caveney NA, Ban F, Cherkasov A, Paetzel M, Strynadka NC. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat Commun, 2020; 11:1–9. CrossRef
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 1997; 23:3–25. CrossRef
Luo X-D, Wu S-H, Ma Y-B, Wu D-G. A new triterpenoid from Azadirachta indica. Fitoterapia, 2000; 71:668–72.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem, 1998; 19:1639–62. CrossRef
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem, 2009; 30:2785–91. CrossRef
Neidle S. Design principles for quadruplex-binding small molecules. Therapeutic applications of quadruplex nucleic acids. Academic Press, Boston, MA, pp 151–74, 2012. CrossRef
Nicola M, Alsafi Z, Sohrabi C, Kerwan A, Al-Jabir A, Iosifidis C, Agha M, Agha R. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int J Surg, 2020; 78:185–93. CrossRef
Ouassou H, Kharchoufa L, Bouhrim M, Daoudi NE, Imtara H, Bencheikh N, ELbouzidi A, Bnouham M. The pathogenesis of coronavirus disease 2019 (COVID-19): evaluation and prevention. J Immunol Res, 2020; 2020:1357983. CrossRef
Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys, 1981; 52:7182–90. CrossRef
Passos MS, Carvalho AR, Boeno SI, Virgens LL, Calixto SD, Ventura TLB, Lassounskaia E, Braz-Filho R, Curcino Vieira IJ. Terpenoids isolated from Azadirachta indica roots and biological activities. Rev Bras Farmacogn, 2019; 29:40–5. CrossRef
Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem, 2015; 58:4066–72. CrossRef
Prieto-Martínez FD, Arciniega M, Medina-Franco JL. Molecular docking: current advances and challenges. TIP Rev Espec En Ciencias Químico-Biológicas, 2018; 21:65–87. CrossRef
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model, 1999; 17:57–61.
Santos LHS, Ferreira RS, Caffarena ER. Integrating molecular docking and molecular dynamics simulations. Methods Mol Biol, 2019; 2053:13–34. CrossRef
Sargsyan K, Grauffel C, Lim C. How molecular size impacts RMSD applications in molecular dynamics simulations. J Chem Theory Comput, 2017; 13:1518–24. CrossRef
Sarkar S, Singh RP, Bhattacharya G. Exploring the role of Azadirachta indica (neem) and its active compounds in the regulation of biological pathways: an update on molecular approach. 3 Biotech, 2021; 11:1–12. CrossRef
Sharon SFB. Molecular docking of selected bioactive compounds from Azadirachta indica for the inhibition of COVID19 protease. Int J Pharm Pharm Sci, 2020; 12:71–7. CrossRef
Siddiqui BS, Ali ST, Rasheed M, Kardar MN. Chemical constituents of the flowers of Azadirachta indica. Helv Chim Acta, 2003; 86:2787–96. CrossRef
Siddiqui S, Faizi S, Siddiqui BS. Constituents of Azadirachta indica: isolation and structure elucidation of a new antibacterial tetranortriterpenoid, mahmoodin, and a new protolimonoid, naheedin. J Nat Prod, 1992; 55:303–10. CrossRef
Singh B, Sharma RA. Secondary metabolites of medicinal plants: ethnopharmacological properties, biological activity and production strategies. John Wiley & Sons, Weinheim, Germany, 2020. CrossRef
Singh KK. Neem, a treatise. IK International Pvt Ltd, New Delhi, India, 2009.
Souza PCT, Limongelli V, Wu S, Marrink SJ, Monticelli L. Perspectives on high-throughput ligand/protein docking with Martini MD simulations. Front Mol Biosci, 2021:199:657222. CrossRef
Stewart JJP. MOPAC2016. Stewart Computational Chemistry, Colorado Springs, CO, 2016. Available via http://openmopac.net/ (Accessed 2 September 2021).
Suárez-Castro A, Valle-Sánchez M, Cortés-García CJ, Chacón-García L. Molecular docking in halogen bonding. Molecular docking. IntechOpen, Paris, France, 2018.
Suárez D, Díaz N. SARS-CoV-2 main protease: a molecular dynamics study. J Chem Inf Model, 2020; 60:5815–31. CrossRef
Sulimov A, Kutov D, Gribkova A, Ilin I, Tashchilova A, Sulimov V. Search for approaches to supercomputer quantum-chemical docking. Russian supercomputing days. Springer, Cham, Switzerland, pp 363–78, 2019. CrossRef
Sulimov A, Kutov D, Ilin I, Sulimov V. Quantum-chemical quasi-docking for molecular dynamics calculations. Nanomaterials, 2022; 12:274. CrossRef
Tallei TE, Tumilaar SG, Niode NJ, Kepel BJ, Idroes R, Effendi Y, Sakib SA, Emran TB. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica (Cairo), 2020; 2020:6307457. CrossRef
Tiwari V, Darmani NA, Yue BYJT, Shukla D. In vitro antiviral activity of neem (Azardirachta indica L.) bark extract against herpes simplex virus type-1 infection. Phyther Res, 2010; 24:1132–40. CrossRef
Tripathy S, Sahu SK. In-silico studies on molecular orbital’s, geometry optimization and molecular docking of curcumin as an antibacterial drug targets FtsZ protein. J Peer Sci, 2018; 1:e1000006.
Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput, 2021; 17:6281–91. CrossRef
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem, 2010; 31:671–90. CrossRef
Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Arthurs S, Colson AB, Freer ST, Larson V, Luty BA, Marrone T, Rose PW. Deciphering common failures in molecular docking of ligand-protein complexes. J Comput Aided Mol Des, 2000; 14:731–51. CrossRef
WHO. WHO Coronavirus (COVID-19) Dashboard 2022. 2022. Available via https://covid19.who.int/ (Accessed 7 December 2022).
Yu W, He X, Vanommeslaeghe K, MacKerell Jr AD. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem, 2012; 33:2451–68. CrossRef
Yusof NA, Kamaruddin S, Abu Bakar FD, Mahadi NM, Abdul Murad AM. Structural and functional insights into TRiC chaperonin from a psychrophilic yeast, Glaciozyma antarctica. Cell Stress Chaperones, 2019; 24:351–68. CrossRef
Zhang L, Dong L, Ming L, Wei M, Li J, Hu R, Yang J. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection during late pregnancy: a report of 18 patients from Wuhan, China. BMC Pregnancy Childbirth, 2020; 20:1–7. CrossRef
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website.