
INTRODUCTION
COVID-19
SARS-CoV-2, the virus that causes COVID-19, also widely dominated as coronavirus, was ubiquitously spread in late 2019 and caused a global epidemic in 2021–2022. Governments confronted the viral outbreak with ordered worldwide country-level lockdowns [1].
Coronaviruses are a large group of RNA viruses in the Coronaviridae family. These viruses are notable for their impact on human health, generating a variety of respiratory infections, such as the current COVID-19 pandemic, and previous outbreaks, such as SARS and MERS. They consist of four distinct subcategories: alpha, beta, gamma, and delta. COVID-19 is a member of betacoronavirus isolated from animals such as pigs and birds [2]. COVID-19 virus has a globular shape with a diameter that ranges between 60 and 140 nm. RNA and phosphorylated nucleocapsid are located in the center of the virus and are surrounded consequently by envelop E protein, spike S protein, and membrane M protein [3]. A high mutation rate in the COVID-19 encoding system is responsible for the variation in S protein structure, which provokes various versions of the virus that vary in terms of infection aggressiveness and host response as symptoms and incubation duration [4]. According to recent data, SARS-CoV-2 is primarily transmitted from person to person. Indeed, the transmission of COVID-19 was reported to be either through direct contact with an infected patient within a conversational distance (1 meter or less) or by aerosols during coughing or sneezing [5,6]. Also, contact with surfaces that are contaminated with the infectious particles and then having direct contact with the eyes, nose, and/or mouth implications could be one of the transmitted routes [5].
COVID-19 infection among mammals is associated with the occurrence of several symptoms that can be classified based on their levels of severity ranging from asymptomatic to severe symptoms [7,8]. Fever, cough, tiredness, and loss of taste or smell can be normal symptoms. However, severe symptoms sought in COVID-19 patients include breathing difficulty or shortness, loss of mobility, and/or chest pain [9]. COVID-19 patients with chronic disorders showed a higher tendency to accuse severe infections that may eventually lead to death. It is worth mentioning that COVID-19 patients have a historically high risk of morbidity, which is primarily caused by pneumonia and shortness of breath [10,11]. According to the World Health Organization’s (WHO) latest statistics, 6.817 million deaths were reported from the beginning of the pandemic until the end of January 2023 [12]. In addition, the fatality rate changed during the pandemic from 2.1% in January 2022, reaching its peak at 9.6% in April 2020, and a decline to 8% recorded in January 2023 [12]. This fate was extremely clear for COVID-19 patients who suffer from cardiovascular diseases (CVD) as they have a fourfold higher risk of death [13]. The majority of fatalities associated with COVID-19 were found to have atrial fibrillation, venous thromboembolism (VTE), and heart failure diagnoses, suggesting that there is an increased risk of cardiovascular consequences after infection with COVID-19 [14]. A further study showed that COVID-19 can endow myocardial injury arrhythmia, acute coronary syndrome, and VTE [15]. These data imply that COVID-19 infection affects the coagulation path for infected patients whether they were already diagnosed by one or more or have acquired the disease post-COVID-19 infection through different means of infection.
Prognostic factors for the severity of COVID-19 infections
The severity of symptoms is attributed to several factors, such as ethnicity, age, gender, medical status, and lifestyle.
Ethnicity
A meta-analysis of the mortality cases was registered in all pandemic countries around the world; the data have revealed that death rates vary according to several factors, such as ethnicity. Indeed, the death rate per ethnicity varies among different ethnicities, where it ranked from highest to lowest as follows: White, Hispanic, and Black for all age categories. Data also revealed that Europe had the highest death rate [16]. Contrarily, recent studies found that Black people had a threefold higher mortality rate from COVID-19 than others. In addition, Hispanic individuals were almost twice as likely to die from COVID-19 compared to non-Hispanic white individuals [17,18]. Moreover, a study shows that Middle Eastern and North African individuals may be at a higher risk of serious disease and mortality from COVID-19 than other populations [19]. Socioeconomic status also plays a role in COVID-19 outcomes. In this context, it was previously reported that the case fatality rate was 5.0% for high-income nations compared to 2.8% for low-income nations [20]. Black and Hispanic populations are more likely to be essential workers and have less access to resources such as paid leave and remote work options, which puts them at higher risk of exposure to the virus. These groups of populations are also more likely to live in poverty and lack resources such as transportation, which can limit their access to healthcare and COVID-19 testing.
AGE
The Centre for Disease Control and Prevention claims that the COVID-19 death rate is the highest among people aged 85 and older. Indeed, reports revealed 1,130 deaths per 100,000 individuals in this age group. This is followed by those aged 65–84 years, with a death rate of approximately 460 deaths per 100,000 individuals. The death rate for the group aged 55–64 years is approximately 140 deaths per 100,000 individuals. These data show that the death rate decreases as age decreases, but severe illness and death can still occur in younger people, especially those with underlying health conditions [21].
Gender
The death rate of COVID-19 has been observed to differ between males and females. Data show that males have a higher death rate than [22]. For example, a study in the United States found that the death rate for male COVID-19 patients was 1.9 times higher than that for female patients [23]. A further study from Jordan shows the death rate is increased in males compared to that in females [24]. Differences in immune response, underlying health conditions, and behavioral factors such as smoking could be explanations. Further research is needed to fully understand the relationship between gender and the death rate of COVID-19 and to develop strategies to mitigate the risks for male patients [25].
IMMUNOLOGY OF COVID-19
Innate immunity: the first line of defense against COVID-19
The body response during COVID-19 infection is initiated by inducing endocytosis of the virus through angiotensin-converting enzyme 2 (ACE2) receptors that are allocated on the surface of epithelial and endothelial cells of the alveoli in the pleural tissue [26,27]. During the COVID-19 virus invasion, the innate immune system acts as the first line of defense against this infection. The Spike (S) protein of the virus binds firmly with ACE2 of epithelial and endothelial cells. In that manner, the viral pathogen-associated molecular patterns (PAMPs) of COVID-19 alert the endosomal pattern recognition receptors (PRRs), such as toll-like receptors, to trigger the immune system to activate the downstream cascades. Consequently, intracellular cascade activates the expression of specific transcription factors like nuclear factor-kappa B and interferon regulatory factors (IRFs) [28]. This cascade motivates the manufacturing of interferons type I (IFNs) and the pro-inflammatory cytokines IFNs urge the antiviral immune state in infected cells that assist in limiting the viral replication and induces apoptosis to preserve the host from viral distribution. Nevertheless, multiple proteins of COVID-19, such as open reading frame 6 and (ORF3b), have a protective mechanism towards antiviral type I IFN (IFN-I) manufacturing and signaling [28–30]. Consequently, for the temporal suppression of the immune response, an utmost over-activation of immune responses eventually leads to hyperinflammation and consequently organ damage. Therefore, the delay in the IFN-I reaction and ubiquitous viral replication in the host cells induce an over-production of IFN-I that could exacerbate hyperactive inflammation combined with a progression of the fatal disease. IFNs stimulate the antiviral immune state in infected cells that limits viral replication and induces apoptosis to protect the host from viral distribution [31].
Immunothrombosis is a process in which the innate immune system’s interaction with the coagulation process is thought to be dysregulated, resulting in localized and/or systemic coagulopathy [32]. The detection of PAMPs and damage-associated molecular patterns (DAMPs) by monocytes expressing PRRs leads to an increase in tissue factor (TF) expression. The extrinsic pathway of coagulation is then activated by TF activation [33], and neutrophil extracellular traps (NETs), which are made of histones that have been acetylated and neutrophil-derived DNA, are also released by active neutrophils. Although NETs are efficient in capturing and eliminating invasive microorganisms, they can also trigger a strong procoagulant response. The complicated pathophysiology seen in severe cases of COVID-19 is a result of the complex interaction between the immune system and coagulation pathways [33]. However, NETs can solely bind to TF to activate the extrinsic coagulation pathway leading to disseminated coagulation in the cardiovascular system. Moreover, the coagulation process is not only dependent on the activation of the extrinsic pathway; factor XII activation can encourage the activation of the intrinsic coagulation pathway. Frequently, in COVID-19, NETs have been seen aggregating with platelets and they may contribute to the severity of the disease [1,34,35].
Adaptive immunity
During COVID-19 infection, the host’s adaptive immunity is ultimately responsible for the virus elimination, B-cell will be activated through different signaling pathways to produce antibodies that neutralize the viral cells and additionally, cytotoxic T-cells will be activated to execute the humeral infected cells [36]. In general, blood lymphopenia is a key indicator of COVID-19 infection; it reduces the number of CD4 + T-cells, CD8 + T-cells, and B-cells [37]. Lymphopenia may occur due to the drop in IFN-I in the innate immune response or due to direct COVID-19 infection of T-cells [38]. However, other factors that contribute to lymphopenia include lymphocyte apoptosis and pyroptosis, lymphocyte sequestration in the lungs, bone marrow hematopoiesis, and virus-induced tissue damage of lymphatic organs [39–41]. COVID-19 directly affects the spleen and lymph nodes by causing splenic white pulp atrophy and lymph node structural disruption, implying that direct COVID-19 cytotoxicity in lymphatic organs may hamper the adaptive immune response in COVID-19 infections [42–44].
Cytokines storm
In many clinical studies that evolved during the pandemic, it was clearly observed that infected patients’ blood analysis inflammatory mediators are at elevated levels, including each cytokine and chemokine [45]. In other words, COVID-19 causes inflammatory disorders in pleural tissue, especially in chemokines levels concomitant with lymphocytopenia and high level of C reactive protein in severely ill patients [26]. This observation can be sought through computerized tomography scans of patients’ lungs, as it revealed a disseminated intrapleural hematoma [37,46]. An interesting finding is that IL-6 is highly correlated to COVID-19 fatality; IL-6 undergoes an amplification cascade that eventually leads to acute respiratory distress syndrome [47,48]. Infected host cells bind with COVID-19 receptors through angiotensin-converting enzyme II (ACE2) that will eventually activate the NF-κB pathway, which triggers IL-6 production [26]. Combined with IL-6 production, a positive feedback loop is activated in the nonimmune cells, thus transducing the effect for IL-6 production. In severe cases, a noticeable increase of CCR6 + Th17 cells and a strong killing ability of CD8 + T cells can be seen in the lungs when using lung scans. At the same time, levels of IL-10 remain unchanged. Notably, both NK cells and CD8 + T cells show signs of weakness in their normal function. In terms of organs, there is a connection between COVID-19 patients’ spleen activity and lymph nodes. This results in the destruction of CD169 + macrophages in both spleens and lymph nodes, causing harm to both lymphoid and splenic tissues. CD169 + macrophages also play a role in increasing levels of Fas, which triggers activation-induced cell death of IL-6 and IL-10 [46].
HEMATOLOGY: COAGULATION CASCADE
Hemostasis is the cascade and events of forming blood coagulations due to sudden injury in the vessels or tissues. Along with this process, there are several clinical symptoms and laboratory evidence for the clots formed, thus increasing the risk of CVD [49]. As shown in Figure 1, fibrinogen and D-dimer are considered the first indicators utilized depending on the type of CVD being investigated. When an internal injury occurs, fibrinogen undergoes a polymerization process where the thrombin enzyme in the plasma catalyzes the process to convert fibrinogen into fibrin. Fibrin is a cross-linked protein structurally more complex than fibrinogen. The enzyme that works against forming clots is called plasmin. It works to disassemble these clots into several byproducts that vary in size and are then called fibrin degradation products (FDP), and one of these products is the D-dimer. Fibrinogen or factor I is a type of protein secreted by the liver. Its function is to normalize the clotting process in the bloodstream [50]. When the fibrinogen concentrations are lowered in the blood, it can be an indicator of abnormal clotting activity, which means that the body is forming clots over normal rates [51]. Measuring fibrinogen’s usually used to detect and treat possible bleeding clotting risks that cause CVDs such as disseminated intravascular coagulation and arterial disease [52].
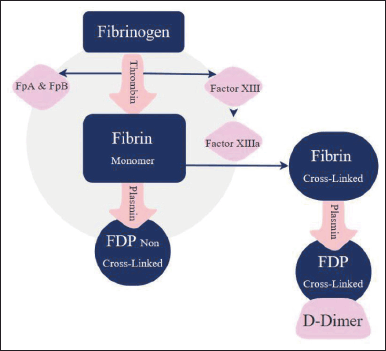 | Figure 1. Fibrinogen degradation [11]. [Click here to view] |
Figure 2 shows the two coagulation pathways; extrinsic and intrinsic. The extrinsic pathway takes place outside the blood and the intrinsic inside it. Each protein in these two pathways is important to proceed in the process of fragmenting produced clots. Any slight change due to genetic mutation will affect the whole process making the individual vulnerable to one CVD, in the worst cases may be morbidity [53]
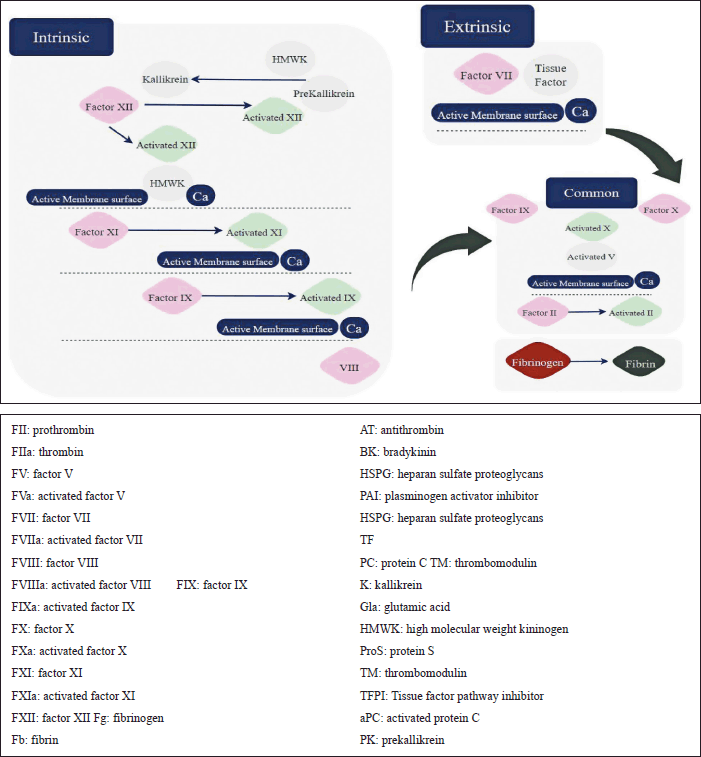 | Figure 2. Coagulation pathway: extrinsic and intrinsic. [Click here to view] |
D-DIMER
D-dimer is formed due to the degradation of cross-linked fibrin [54]. It is a small fragment generated when the fibrin clot is broken down by plasmin, a protein that helps dissolve clots. High D-dimer levels in the blood indicate that the coagulation system is activated and that the fibrin clot is degrading. Thus, D-dimer testing can help diagnose deep vein thrombosis (DVT) [55]. Elevated D-dimer levels have been seen in a considerable proportion of COVID-19 patients. This elevation is assumed to be due to coagulation system activation, a common feature of the disease. The coagulopathy observed in COVID-19 patients is caused by the virus’s ability to directly affect the blood vessels and the immune system’s over-reactive response to the virus, leading to increased clotting activity and higher levels of D-dimer [56,57]. The relationship between D-dimer and COVID-19 is particularly important in severe cases where elevated levels of D-dimer can contribute to the development of VTE and other clotting disorders. Moreover, elevated D-dimer levels have also been linked to an increase in hospitalization risk, ICU admission, and death in COVID-19 patients, highlighting the importance of monitoring D-dimer levels in these individuals [58].
Genetic mutations affecting D-dimer functionality
The normal coagulation pathway will be harmed if the genetic substance responsible for coagulation proteins is changed. Fibrinogen is coded by mainly three genes for each component. These genes can be found in a region of ~50 kb on chromosome 4q28–q31. In genome-wide association study, single nucleotide polymorphisms (SNPs) that strongly affect D-dimers are mainly found in fibrinogen gamma, fibrinogen alpha, and factor V, and other genes such as MTHFR, KLKB1, KNG1, F2, F8, and F11 also take place in the process [59]. Three recommended SNPs-ID responsible for D-dimer elevated levels are shown in Table 1. However, other SNPs such as AC093117.1 and Z99572.1 play a role in type 2 diabetes and end-stage coagulation and coagulants, factors II and V, respectively.
A) FACTOR III
The F3 locus, also known as TF or thromboplastin, which is located 46.0 kb upstream from the start of transcription in the hypothesized regulatory region of F3, has been discovered as a source of inherited thrombophilia, a condition characterized by an increased tendency to form blood clots [60]. Factor III is a major component of the coagulation cascade and the primary activator of the extrinsic pathway. Factor III, also known as TF or thromboplastin, is normally present in low levels in subendothelial tissue, but it is exposed to circulating blood upon vascular injury [61], and concomitantly factor VII (FVII) is activated when TF forms a complex with FVII to become its active form, FVIIa. Subsequently, factor X (FX) is activated by this complex, known as TF-FVIIa, by cleaving it to its active form, FXa [62], to form the prothrombinase complex. Later on, the active factor Xa (FXa) joins with factor Va (FVa), a cofactor, and calcium ions (Ca2+) to activate the pathway. The prothrombinase complex is essential for converting prothrombin (factor II) to thrombin (factor IIa) via a restricted proteolysis process [63]. The serine protease thrombin plays several purposes in the coagulation cascade. To generate an insoluble fibrin clot, it breaks down fibrinogen (factor I) into fibrin monomers Liu and Cao, [64]. In addition, thrombin leads to platelet activation and improves coagulation by activating factors such as factors V, VIII, and XIII [65]. The TF mechanism initiates the coagulation cascade and leads to the production of a stable fibrin clot in response to vascular damage. The dysregulation of this process, which is closely regulated and involves a number of proteases, cofactors, and their interactions, can have important clinical complications for thrombotic diseases [66]. F3 influences D-dimer levels by triggering the extrinsic pathway, leading to coagulation alterations and fibrinolysis, ultimately affecting the quantity of shed D-dimer fragments. There is a growing curiosity about the involvement of TFs in initiating hemostasis and its impact on arterial and venous thrombosis, inflammation, and the growth and spread of tumors [67]. Also, in [68], a study showed that individuals with F3 gene mutations had significantly higher levels of D-dimer compared to those without the mutations (p < 0.001). The mean D-dimer levels in the group with F3 gene mutations were found to be 2.0 ng/ml, while the mean D-dimer levels in the group without the mutations were found to be 1.5 ng/ml.
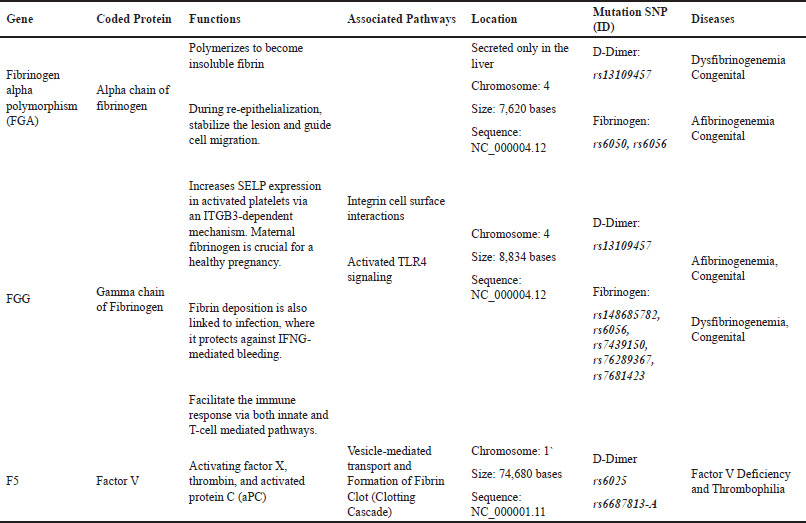 | Table 1. Description of main genes affecting DD and fibrinogen. [Click here to view] |
B) Factor V Leiden (FVL)
The formation of blood clots depends greatly on factor V, a crucial component of the coagulation cascade. In the coagulation cascade, factor V activates and interacts with other factors to produce thrombin and fibrin [69]. In the normal coagulation process, factor V is converted from the inactive form (factor V) to the active form (factor Va) by the action of thrombin, which is produced by the activation of factor X. Once active, factor Va works as a cofactor for the enzyme factor Xa, boosting its ability to convert prothrombin to thrombin. Then, fibrinogen is converted into fibrin monomers by the action of thrombin [70], activated protein C (APC), a natural anticoagulant, controls factor V as well. APC inactivates factor Va by cleaving it at certain locations, preventing excessive clot formation. This regulatory system aids in regulating the ratio of clotting to clot disintegration [71]. FVL is a genetic mutation that causes an arginine residue at position 506 of the factor V protein to be replaced with a glutamine residue. This mutation makes activated protein C, a critical regulator of the coagulation system, resistant to inactivation. As a result, people with the FVL mutation are more likely to develop thrombosis or the formation of blood clots [72]. A study conducted by [73] looked at the relationship between FVL and D-dimer levels in a large population-based cohort. The results showed that individuals with the FVL mutation had significantly higher levels of D-dimer compared to individuals without the mutation. This suggests that the FVL mutation may be associated with an increased risk of VTE and elevated D-dimer levels. A study conducted by [74,75] evaluated the relationship between FVL and D-dimer levels in patients with DVT. The results showed that individuals with the FVL mutation had higher D-dimer levels than those without the mutation. This further supports the association between the FVL mutation and elevated levels of D-dimer.
C) Fibrinogen alpha chain
Fibrinogen is a glycoprotein that is essential for the process of blood clotting. It is composed of three chains: alpha, beta, and gamma. The alpha chain of fibrinogen has been the focus of recent research due to its role in the development of DVT, a condition that results from the formation of blood clots in the deep veins of the legs [76]. The fibrinogen chain is essential in the coagulation cascade, which is initiated by vascular damage. The activation of clotting factors, particularly factor Xa, results in the production of thrombin as a part of the cascade. As a serine protease, thrombin breaks down certain peptide connections in fibrinogen, releasing the fibrinopeptides A and B [77]. This cleavage exposes binding sites on fibrinogen, allowing individual fibrinogen molecules to join and produce fibrin monomers. The subsequent polymerization of these monomers, assisted by factor XIIIa, results in the formation of a network of lengthy fibrin strands [79]. The fibrin network serves as the structure that supports a blood clot. Cross-linking, which is also made possible by factor XIIIa, stabilizes the clot by forming a stable three-dimensional clot. This process involves the integration of numerous clotting factors, enzymes, and cofactors [80]. Genetic variations in the alpha chain of fibrinogen can affect its function and contribute to the risk of developing DVT. For example, the FGA gene has been identified as a risk factor for DVT. This gene encodes the alpha chain of fibrinogen and variants of this gene have been associated with increased levels of fibrinogen, leading to a greater risk of DVT [81]. In addition, this research has indicated that elevated levels of fibrinogen, specifically the alpha chain, can result in a higher risk of DVT and other thromboembolic events. The alpha chain of fibrinogen has been shown to play a crucial role in the formation and stability of clots, and the presence of genetic variations in this gene may result in increased clotting and a higher risk of DVT [82]. In general, the research indicated that the alpha chain of fibrinogen plays an important role in the development of DVT. In addition, genetic variations in this gene can affect its function and contribute to an increased risk of DVT.
COVID-19 impacting effects on the coagulation cascade
Researchers have studied the relationship between D-dimer values and tried to correlate it to COVID-19 infection; all studies have a consensus that D-dimer levels are high during COVID-19 infection. For example, a study conducted on 191 patients and approximately 28% death rates of the population traced laboratory markers for both survival and non-survival patients and concluded that D-dimer levels are surprisingly high for fatalities. In such a case and after recognizing that D-dimer levels are raised due to COVID-19 infection, one may be concerned about whether COVID-19 infection may cause one or more of the D-dimer mutations [83]. As shown in Figure 3, COVID-19 is a systemic, hypercoagulable disease. The fibrinogen, the source of D-dimer, is secreted in the liver as a response to high IL-1 and IL-6, and when a coagulation activity takes place, increasing fibrinogen levels and thus D-dimer levels, both values are good indicators for coagulation activity related to COVID-19 [84]. COVID-19 morbidities were first justified due to pneumonia and shortness of breath; however, it is important to investigate the role of thrombosis in such cases [85].
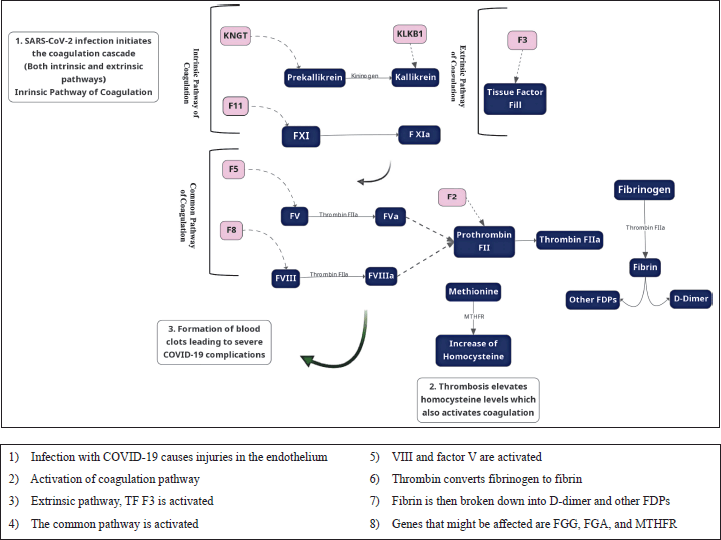 | Figure 3. Molecular link between key genes in coagulation and COVID-19. [Click here to view] |
CONCLUSION
COVID-19 has been observed to exacerbate changes in coagulopathy through various mechanisms. These mechanisms include the activation of both the extrinsic and intrinsic pathways via factor III, factor V, and FGA|FGG. In addition, genetic variations can result in the predicted elevation of D-dimer levels, particularly in elderly individuals and those with age-related influences. Further investigation is needed to fully understand the correlation between D-dimer levels and COVID-19.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11). doi: https://doi.org/10.1172/JCI.INSIGHT.138999
2. Burrell CJ, Howard CR, Murphy FA. History and impact of virology. Fenner and White’s Med Virol 2017:3. doi: https://doi.org/10.1016/B978-0-12-375156-0.00001-1
3. Hussen BM, Sabir DK, Karim Y, Karim KK, Hidayat HJ. Genome sequence analysis of SARS-COV-2 isolated from a COVID-19 patient in Erbil, Iraq. Appl Nanosci (Switzerland) 2022;1:1–7. doi: https://doi.org/10.1007/S13204-021-02300-W/FIGURES/3
4. Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7:1012. doi: https://doi.org/10.1093/NSR/NWAA036
5. Liu J, Liao X, Qian S, Yuan J, Wang F, Liu Y, et al. Community transmission of severe acute respiratory syndrome coronavirus 2, Shenzhen, China, 2020. Emerg Infect Dis. 2020;26:1320–3. doi: https://doi.org/10.3201/EID2606.200239
6. Rothe C, Schunk M, Sothmann P, Bretzel G, Froeschl G, Wallrauch C, et al. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N Engl J Med. 2020;382:970–1. doi: https://doi.org/10.1056/NEJMC2001468
7. Chung MK, Zidar DA, Bristow MR, Cameron SJ, Chan T, Harding CV 3rd, et al. COVID-19 and cardiovascular disease: from bench to bedside. Circ Res. 2021;128:1214–36. doi: https://doi.org/10.1161/CIRCRESAHA.121.317997
8. Rayyan WA. Seroprevalence of SARS-CoV-2 antibodies among Jordanian citizens: a cross-sectional study of the demographic and clinical factors that ameliorate serum IgG concentration. J Appl Pharm Sci. 2022; 12(11):151–6. doi: https://doi.org/10.7324/JAPS.2022.121116
9. Talukder A, Razu SR, Alif SM, Rahman MA, Islam SMS. Association between symptoms and severity of disease in hospitalised novel coronavirus (COVID-19) patients: a systematic review and meta-analysis. J Multidiscip Healthc. 2022;15:1101–10. doi: https://doi.org/10.2147/JMDH.S357867
10. George PM, Barratt SL, Condliffe R, Desai SR, Devaraj A, Forrest I, et al. Respiratory follow-up of patients with COVID-19 pneumonia. Thorax. 2020;75:1009–16. doi: https://doi.org/10.1136/THORAXJNL-2020-215314
11. Rayyan WA, Hazzaa WA, Seder N, Al-Fawares O, Fararjeh AFS. The implications of COVID-19 infection on hematologic parameters and coagulation activity: a review. Biomed Pharmacol J. 2022;15:1837–51. doi: https://doi.org/10.13005/bpj/2522
12. World Health Organisation. No Title 2022.
13. Cordero A, Santos García-Gallego C, Bertomeu-González V, Fácila L, Rodríguez-Mañero M, Escribano D, et al. Mortality associated with cardiovascular disease in patients with COVID-19. REC: CardioClinics. 2021;56:30–8. doi: https://doi.org/10.1016/j.rccl.2020.10.005
14. Raisi-Estabragh Z, Harvey NC, Petersen SE. Response to: correspondence on ‘cardiovascular disease and mortality sequelae of COVID-19 in the UK biobank’ by Jolobe. Heart. 2023;109:332 LP–333. doi: https://doi.org/10.1136/heartjnl-2022-322124
15. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020;17:543–58. doi: https://doi.org/10.1038/S41569-020-0413-9
16. Noor FM, Islam MM. Prevalence and associated risk factors of mortality among COVID-19 patients: a meta-analysis. J Community Health. 2020;45:1270–82. doi: https://doi.org/10.1007/S10900-020-00920-X/TABLES/3
17. Webb Hooper M, Nápoles AM, Pérez-Stable EJ. COVID-19 and racial/ethnic disparities. JAMA. 2020;323:2466–7. doi: https://doi.org/10.1001/jama.2020.8598
18. Aburto M, Tilstra AM, Floridi G, Dowd JB. Significant impacts of the COVID-19 pandemic on race/ethnic differences in US mortality. Proc Natl Acad Sci U S A. 2022;119(35):1–9. doi: https://doi.org/10.1073/pnas.2205813119/-/DCSupplemental.Published
19. Boufkhed S, Harding R, Kutluk T, Husseini A, Pourghazian N, Shamieh O. What is the preparedness and capacity of palliative care services in middle-eastern and north African countries to respond to COVID-19? a rapid survey. J Pain Symptom Manage. 2021;61:e13–50. doi: https://doi.org/10.1016/j.jpainsymman.2020.10.025
20. Sreedharan J, Nair SC, Muttappallymyalil J, Gopakumar A, Eapen NT, Satish KP, et al. Case fatality rates of COVID-19 across the globe: are the current draconian measures justified? Z Gesundh Wiss. 2022;30:2575–83. doi: https://doi.org/10.1007/s10389-021-01491-4
21. Tejada-Vera B, Kramarow EA. COVID-19 Mortality in adults aged 65 and over: United States, 2020. NCHS Data Brief. 2022:1–8.
22. Jin J-M, Bai P, He W, Wu F, Liu X-F, Han D-M, et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. 2020;8:152. doi: https://doi.org/10.3389/fpubh.2020.00152
23. Gold JAW, Wong KK, Szablewski CM, Patel PR, Rossow J, da Silva J, et al. Characteristics and clinical outcomes of adult patients hospitalized with COVID-19 — georgia, march 2020. MMWR Morb Mortal Wkly Rep. 2020;69:545–50. doi: https://doi.org/10.15585/MMWR.MM6918E1
24. Khader Y, Al Nsour M. Excess mortality during the COVID-19 pandemic in Jordan: secondary data analysis. JMIR Public Health Surveill. 2021;7:e32559. doi: https://doi.org/10.2196/32559
25. Danielsen AC, Lee KM, Boulicault M, Rushovich T, Gompers A, Tarrant A, et al. Sex disparities in COVID-19 outcomes in the United States: quantifying and contextualizing variation. Soc Sci Med. 2022;294:114716. doi: https://doi.org/10.1016/j.socscimed.2022.114716
26. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11:875–9. doi: https://doi.org/10.1038/nm1267
27. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. doi: https://doi.org/10.1016/J.EJIM.2020.04.037
28. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: https://doi.org/10.1016/J.CELL.2006.02.015
29. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32. doi: https://doi.org/10.1016/J.CELREP.2020.108185
30. Xia H, Cao Z, Xie X, Zhang X, Chen JYC, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33(1). doi: https://doi.org/10.1016/J.CELREP.2020.108234
31. Tian W, Zhang N, Jin R, Feng Y, Wang S, Gao S, et al. Immune suppression in the early stage of COVID-19 disease. Nat Commun. 2020;11(1):5859. doi: https://doi.org/10.1038/S41467-020-19706-9
32. Nakazawa D, Ishizu A. Immunothrombosis in severe COVID-19. EbioMedicine. 2020;59:102942. doi: https://doi.org/10.1016/J.EBIOM.2020.102942
33. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45. doi: https://doi.org/10.1038/NRI3345
34. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020;1:e290–9. doi: https://doi.org/10.1016/S2666-5247(20)30144-0
35. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169. doi: https://doi.org/10.1182/BLOOD.2020007008
36. Sette A, Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184:861–80. doi: https://doi.org/10.1016/j.cell.2021.01.007
37. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620–9. doi: https://doi.org/10.1172/JCI137244
38. Lo Presti E, Dieli F, Meraviglia S. Lymphopenia in COVID-19: γδ T Cells-based therapeutic opportunities. Vaccines (Basel). 2021;9(6):562. doi: https://doi.org/10.3390/vaccines9060562
39. Moon C. Fighting COVID-19 exhausts T cells. Nat Rev Immunol. 2020;20:277. doi: https://doi.org/10.1038/S41577-020-0304-7
40. Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat Commun. 2020;11(1):5086. doi: https://doi.org/10.1038/S41467-020-18854-2
41. Janssen NAF, Grondman I, De Nooijer AH, Boahen CK, Koeken VACM, Matzaraki V, et al. Dysregulated innate and adaptive immune responses discriminate disease severity in COVID-19. J Infect Dis. 2021;223:1322–33. doi: https://doi.org/10.1093/infdis/jiab065
42. Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. 2020;396:320–32. doi: https://doi.org/10.1016/S0140-6736(20)31305-2
43. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. 2020;183:143–57.e13. doi: https://doi.org/10.1016/J.CELL.2020.08.025
44. Tillett RL, Sevinsky JR, Hartley PD, Kerwin H, Crawford N, Gorzalski A, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis. 2021;21:52–8. doi: https://doi.org/10.1016/S1473-3099(20)30764-7
45. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–74. doi: https://doi.org/10.1038/S41577-020-0311-8
46. Luo M, Liu J, Jiang W, Yue S, Liu H, Wei S. IL-6 and CD8+ T cell counts combined are an early predictor of in-hospital mortality of patients with COVID-19. JCI Insight 2020;5(13):e139024. doi: https://doi.org/10.1172/JCI.INSIGHT.139024
47. Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52:731–3. doi: https://doi.org/10.1016/J.IMMUNI.2020.04.003
48. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020;19(6):102537. doi: https://doi.org/10.1016/J.AUTREV.2020.102537
49. LaPelusa A, Dave HD. Physiology, hemostasis. StatPearls 2021.
50. Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. JThromb Haemost. 2015;13:417–25. doi: https://doi.org/10.1111/jth.12802
51. Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58:515. doi: https://doi.org/10.4103/0019-5049.144643
52. Lowe GDO, Rumley A, Mackie IJ. Plasma fibrinogen. Ann Clin Biochem. 2004;41:430–40. doi: https://doi.org/10.1258/0004563042466884
53. Mahdieh N, Rabbani B. An overview of mutation detection methods in genetic disorders. Iran J Pediatr. 2013;23:375.
54. Deme D, Telekes A. [Prognostic importance of cross-linked fibrin degradation products (D-dimer) in oncology]. Magy Onkol. 2017;61:319–26.
55. Konstantinides SV, Meyer G, Becattini C, Bueno H, Geersing G-J, Harjola V-P, et al. 2019 ESC guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European respiratory society (ERS). Eur Heart J. 2020;41:543–603. doi: https://doi.org/10.1093/eurheartj/ehz405
56. Haematology TL. COVID-19 coagulopathy: an evolving story. Lancet Haematol. 2020;7:e425. doi: https://doi.org/10.1016/S2352-3026(20)30151-4
57. Logothetis CN, Weppelmann TA, Jordan A, Hanna C, Zhang S, Charkowick S, et al. D-Dimer testing for the exclusion of pulmonary embolism among hospitalized patients with COVID-19. JAMA Netw Open. 2021;4:e2128802. doi: https://doi.org/10.1001/jamanetworkopen.2021.28802
58. Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost. 2020;18:2103–9. doi: https://doi.org/10.1111/jth.14975
59. Bosso M, Thanaraj TA, Abu-Farha M, Alanbaei M, Abubaker J, Al-Mulla F. The Two faces of ACE2: the role of ACE2 receptor and its polymorphisms in hypertension and COVID-19. Mol Ther Methods Clin Dev. 2020;18:321–7. doi: https://doi.org/10.1016/J.OMTM.2020.06.017
60. Smith NL, Huffman JE, Strachan DP, Huang J, Dehghan A, Trompet S, et al Genetic predictors of fibrin D-dimer levels in healthy adults. Circulation. 2011;123:1864–72. doi: https://doi.org/10.1161/CIRCULATIONAHA.110.009480
61. Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–22. doi: https://doi.org/10.1161/01.ATV.0000130465.23430.74
62. Gailani D, Renné T. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2507–13. doi: https://doi.org/10.1161/ATVBAHA.107.155952
63. Steen M. Factor Va-factor Xa interactions: molecular sites involved in enzyme:cofactor assembly. Scand J Clin Lab Invest Suppl. 2002;237:5–12. doi: https://doi.org/10.1080/003655102762377439
64. Liu W, Cao Y. Tissue engineering technology for tissue repair and regeneration. Compr Biotechnol. 2019:173–201. doi: https://doi.org/10.1016/B978-0-444-64046-8.00300-1
65. Tomaiuolo M, Brass LF, Stalker TJ. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv Cardiol Clin. 2017;6:1. doi: https://doi.org/10.1016/J.ICCL.2016.08.001
66. Unruh D, Mirkov S, Wray B, Drumm M, Lamano J, Li YD, et al. Methylation-dependent tissue factor suppression contributes to the reduced malignancy of idh1-mutant gliomas. Clin Cancer Res. 2019;25:747–59. doi: https://doi.org/10.1158/1078-0432.CCR-18-1222
67. Mackman N. The many faces of tissue factor. J Thromb Haemost. 2009;7:136. doi: https://doi.org/10.1111/J.1538-7836.2009.03368.X
68. Francis CW, Hogg N HJ. Francis CW, Hogg N, Hargrove J, et al. Fibrinogen (Factor III) gene mutations and thrombophilia. 2000;215–20.
69. Smith SA, Travers RJ, Morrissey JH. How it all starts: Initiation of the clotting cascade. Crit Rev Biochem Mol Biol. 2015;50:326–36. doi: https://doi.org/10.3109/10409238.2015.1050550
70. Brown MA, Stenberg LM, Stenflo J. Coagulation Factor Xa. Handbook of Proteolytic Enzymes. 2013;3:2908. doi: https://doi.org/10.1016/B978-0-12-382219-2.00642-6
71. Cramer TJ, Griffin JH, Gale AJ. Factor V Is an Anticoagulant Cofactor for Activated Protein C during Inactivation of Factor Va. Pathophysiol Haemost Thromb. 2010;37:17. doi: https://doi.org/10.1159/000315141
72. Lee S, Lee CH, Seo MS, Yoo J il. Integrative analyses of genes about venous thromboembolism: An umbrella review of systematic reviews and meta-analyses. Medicine. 2022;101:e31162. doi: https://doi.org/10.1097/MD.0000000000031162
73. Meer FJ, Rosendaal FR, de Groot PG, Reitsma PH. D-dimer levels and the risk of venous thrombosis in carriers of the factor V Leiden mutation. Thromb Haemost. 2003;89:487–91.
74. Koster T, Rosendaal FR, Bertina RM, Reitsma PH. Elevated levels of D-dimer are associated with the factor V Leiden mutation. Thrombosis Haemostasis. 2010;103:96–100.
75. Kujovich JL. Factor V Leiden thrombophilia. Genet Med. 2011;13:1–16. doi: https://doi.org/10.1097/GIM.0B013E3181FAA0F2
76. Wallentin L, Hansen ML, Almdahl SM, the WODIT study group. Fibrinogen alpha-chain polymorphism and the risk of venous thromboembolism. results from the western norway diet and health study. Thromb Haemost. 2009Oct;102(4):777–81.
77. Weisel JW, Litvinov RI. Fibrin formation, structure and properties. Subcell Biochem. 2017;82:405. doi: https://doi.org/10.1007/978-3-319-49674-0_13
78. Weisel JW, Litvinov RI. Fibrin formation, structure and properties. Subcell Biochem. 2017;82:405–56. doi: https://doi.org/10.1007/978-3-319-49674-0_13
79. Marín-García J. Molecular basis of lipoprotein disorders, atherogenesis, and thrombosis. Post-Genomic Cardiol. 2007;1:211–60. https://doi.org/10.1016/B978-012373698-7/50008-5
80. Aleman MM, Walton BL, Byrnes JR, Wolberg AS. Fibrinogen and red blood cells in venous thrombosis. Thromb Res. 2014;133:S38–40. doi: https://doi.org/10.1016/J.THROMRES.2014.03.017
81. Righini M, the EINDT study group. The fibrinogen alpha polymorphism and risk of venous thromboembolism: a meta-analysis. Thromb Haemost. 2006;96(4):599–604.
82. Manco L, Limana L, Landi G. The association of fibrinogen alpha chain with deep vein thrombosis. vol. Jul;154. 2017.
83. Hayiroglu MI, Cinar T, Tekkesin AI. Fibrinogen and D-dimer variances and anticoagulation recommendations in COVID-19: current literature review. Rev Assoc Med Bras. (1992) 2020;66:842–. doi: https://doi.org/10.1590/1806-9282.66.6.842
84. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. 2009;145:24–33. doi: https://doi.org/10.1111/J.1365-2141.2009.07600.X
85. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. doi: https://doi.org/10.1016/s0140-6736(20)30566-3