
INTRODUCTION
Natural products can serve as budding resources for the development of anti-inflammatory drugs due to better pharmacological activities and lower toxicity (Deng et al., 2010). Presently, almost all drugs are derived from plant origins. Plant constituents are attractive agents with low cost, effectiveness, and biocompatibility, which makes phytochemicals a prominent cause for the development and identification of the lead compounds that provide support to the pharmacological activity of existing drugs (Samuelsen, 2000). Plant extracts have been reported with enormous biological activities like anti-inflammatory, antioxidant, wound healing activity, weak antibiotic, analgesic, immunomodulating, and antiulcerogenic activity (Mozaffarian, 2012)
Plantago major is a perennial plant that belongs to the family Plantaginaceae. It consists of biologically potent compounds like alkaloids, polysaccharides, flavonoids, iridoid glycosides, lipids, caffeic acid derivatives, terpenoids, and organic acids (Samuelsen, 2000) (Fig. 1). These compounds can be found in almost all parts of the plant such as the seeds, leaves, ?ower, and roots (Adom et al., 2017; Wang et al., 2015). Earlier studies reported that P. major is used in different parts of the world for the treatment of numerous conditions like skin diseases, infectious diseases, digestive problems, respiratory abnormalities, glitches related to reproduction, circulation, tumors, and inflammation (Azab et al., 2016). Owing to the tradition of employing P. major for wound healing, it is worth exploring this plant further for anti-inflammatory activity (Mozaffarian, 2012; Samuelsen, 2000)
In recent years, great advancement has been made in the expansion and resolution mechanisms of chronic inflammatory diseases, and the use of phytoconstituents to lighten inflammatory diseases (Isailovic et al., 2015). We are persistent in exploring the anti-inflammatory targets in the context of chronic inflammation, establishing a screening technique, and identification of potent leads with strong anti-inflammatory activity (Deng et al., 2010; Newman and Cragg, 2007)
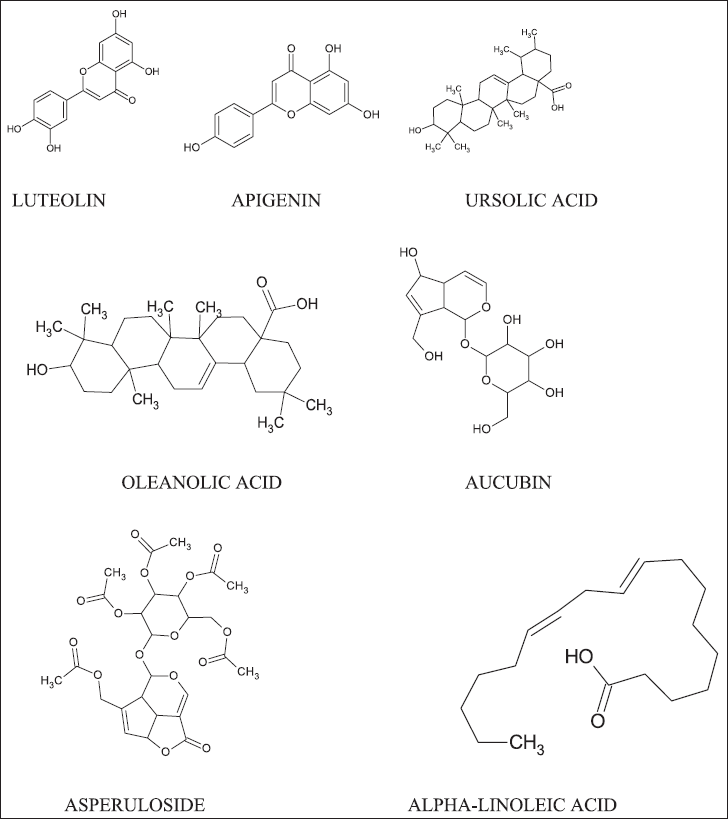 | Figure 1. Major constituents of P. major. [Click here to view] |
Some of the structures of the major constituent are illustrated in Figure 1.
Molecular modeling (MM) is a significant improvement in the design and discovery of new leads (Langer and Hoffmann, 2001). At present, MM is considered a necessary tool in drug discovery and optimizing the prevailing prototypes and the rational design of lead candidates, in which virtual screening plays a prominent role (Lionta et al., 2014). Due to that, numerous procedures, such as the pharmacophore model development, molecular dynamics, etc., have been recognized (Saikiran Reddy et al., 2018).
The ligand-based pharmacophore method is used to develop a pharmacophore model. The current research work encompasses various phytoconstituents to identify and design new molecules with potent anti-inflammatory activity. Phytoconstituents are an attractive source in developing the lead molecules for various diseases.
The basis of the perception is that molecules that share few structural similarities may possess the same activity. Some approaches use existing active ligands as a query to retrieve structurally analogous compounds from huge databases (Lionta et al., 2014).
In present computational chemistry, the vital topographies of ligands with the same pharmacological activity were well-defined using pharmacophore (Kaserer et al., 2015). It is defined as the three-dimensional alignment of features required for a ligand to interact with a particular protein. These particular traits are substantial to accomplish the finest pharmacophore model. In this research, PharmaGist, a web-based software that is a ligand-based method was used to build a pharmacophore model (Schneidman-Duhovny et al., 2008; Usha, 2016). Molecular interactions and their predictions are a prominent step in rational drug design. The spatial alignment of features that is crucial for a molecule to interact with a definite target is a pharmacophore. The ligand-based screening method is employed here (Dror et al., 2009).
The screening process includes choosing the finest pharmacophore employing the Pharmit server’s inbuilt database (Sunseri and Koes, 2016) and validating through the docking studies. Pharmit offers an online, user-friendly environment for the virtual screening of large databases using pharmacophores, energy minimization, and molecular shape. It also provides easy access to large chemical datasets. Docking studies were performed to identify the screened molecules’ affinity toward the protein target, which is one of the major steps in the rational drug design process (Saikiran Reddy et al., 2018).
MATERIALS AND METHODS
Selection of target protein
The 3D structure of Cyclooxygenase-2 (PDB ID: 4COX) was downloaded from Protein Data Bank (https://www.rcsb.org) (Kurumbail et al., 1996). The crystal resolution of 4COX is 2.90 A°.
Ligand selection for pharmacophore modeling
The 3D structures of selected ligands were retrieved from PubChem in SDF format (Kim et al., 2016). Later, the Open Babel server was used to convert all five ligand structures into (Fig. 2) mol2 format (O’Boyle et al., 2011). All the phytoconstituents having a good anti-inflammatory activity (aucubin, luteolin, alpha-linoleic acid, oleanolic acid, ursolic acid) were used to build a pharmacophore model with “PharmaGist webserver.”
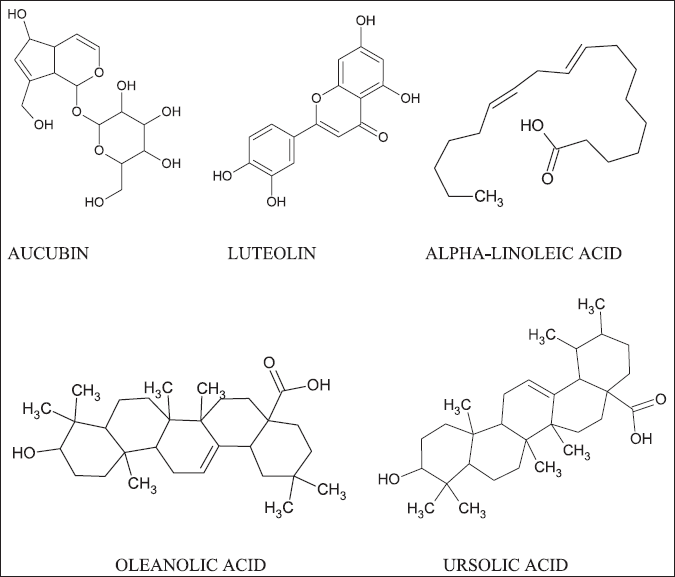 | Figure 2. The structures of five plant (P. Major) components having anti-inflammatory activity. [Click here to view] |
PharmaGist
The best open-source web server used for searching a pharmacophore using a group of ligands that bind to a protein is PharmaGist. For every input ligand, this web server lists the number of atoms and three-dimensional and physicochemical features like aromatic rings, hydrophobic regions, hydrogen bond donors, and hydrogen bond acceptors as given in Table 1. This method expeditiously searches for all possible pharmacophores, generates three-dimensional visualization of detected pharmacophores by multiple alignments of input ligands, and executes virtual screening based on the detected pharmacophore model.
Virtual screening
The built pharmacophore model is used as a query in Pharmit to screen libraries of small molecules. Load pharmacophore features and selects a database for screening (e.g., National Cancer Institute (NCI) Open chemical repository database). The number of hits was displayed, to reduce the hits. The flow chart is depicted in Figure 3.
Pharmit
It is an online open-source platform for performing structure-based virtual screening with pharmacophore and shape queries (http://pharmit.csb.pitt.edu). It allows users to interactively search libraries of millions of compounds as part of structure-based drug discovery. It enables us to explore ligands grounded on their chemical and structural similarity to other ligands. The compounds which exhibited extreme similarity to query pharmacophore were filtered.
Molecular docking studies
Dataset ligands and ligand optimization
ACD/Chemsketch was used to generate the 2D ligand structures. The generated ligands were subjected to 3D optimization and saved in Molecular Dynamics Language Molfile format. Later, by using Open Babel, optimized ligands were converted to a Protein Data Bank, Partial Charge (Q), & Atom Type (T) format (PDBQT) file format.
The downloaded crystal structure of the protein (PDB ID: 4COX) was optimized by removing water molecules, hydrogen atoms were added to satisfy the valences, charges were included, further missing amino acids were added to stabilize side chains, and energy of the complete structure was minimized with AUTODOCK suite of Molecular Graphics Laboratory Tools.
Molecular docking studies were performed with Autodock Vina (Muni Sireesha et al., 2022). A grid was created around the co-crystallized ligand. The coordinates (x = 22.51, y = 22.24, z = 16.199) were produced with the help of Pharmit server (http://pharmit.csb.pitt.edu/). Protein target and ligand PDBQT files were prepared. In the absence of water molecules, docking was performed for all 10 compounds (9 + 1 standard). Subsequently, all the molecules were analyzed and visualized in the discovery studio for the interactions with the active site (MuniSireesha et al., 2021). Further, the efficiency of the binding and its interactions was calculated in terms of dock score, which is a blend of hydrophobic, hydrophilic, metal binding groups, freezing rotatable bond, Vander Waals energy, and polar interactions with receptors.
 | Figure 3. Representation of pharmacophore development. [Click here to view] |
RESULT AND DISCUSSION
Detection of pharmacophore model
PharmaGist helps in detecting pharmacophore, which is the three-dimensional alignment of various topographies that allows a ligand to interact with the protein in its binding pocket. The input of a group of ligands in PharmaGist will highlight Pharmacophore candidates by superimposing the three-dimensional ligand conformations. The selected ligands are phytoconstituents of P. major with anti-inflammatory activity. Further, the ligands were imported into Marvin viewer and saved in Tripos mol2 format. Later, the individual mol2 files are compressed as a zip file and submitted as a query in PharmaGist to identify the pharmacophore candidates as given in Table 2. Based on the score retrieved by the pairwise alignment in the PharmaGist server, the best ligands were selected as mentioned in Table 3. The pairwise alignment and structures of the ligands, the bond angles, and bond distances are given in Figure 4.
Virtual screening
The output pharmacophore model 1 (PM1) obtained as a mol2 file through PharmaGist software which has the highest score of 12.402 was submitted as a query to the Pharmit web server to visualize the alignment of the standard with the ligands (Fig. 5). It is a four-point pharmacophore having features of one hydrogen bond donor and three hydrogen bond acceptors. From over 52,237 ligands, the virtual screening process retrieved 11,341 hits, which are matched with the features in the query pharmacophore model. Then by applying filters of the Lipinski rule, root mean squared distance (RMSD), and rotatable bonds, finally we selected the top-nine hits. Further, these nine hits were subjected to molecular docking analysis to prioritize the best ligand for the anti-inflammatory target.
Molecular docking
Molecular docking was performed with the X-ray crystal structures of the target (PDB ID: 4COX), pivot molecule ursolic acid, and four input ligand molecules (aucubin, luteolin, alpha-linoleic acid, oleanolic acid, ursolic acid). Subsequently, the screened nine hit molecules from the NCI database were sketched in Chemdraw and saved in mol2000 format. All the ligand molecules’ energy was minimized and converted into PDBQT format using the Open Babel server. All the compounds’ low energy conformations were docked into the active site of the target, i.e., 4COX using AUTODOCK VINA. It is a technique for assessing the accuracy of the docking process to determine how closely the lowermost binding configuration is predicted by the object score function.
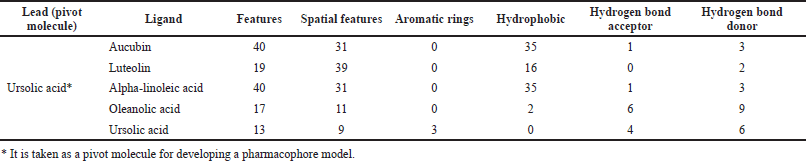 | Table 1. Pharmacophoric characteristics of selected ligands. [Click here to view] |
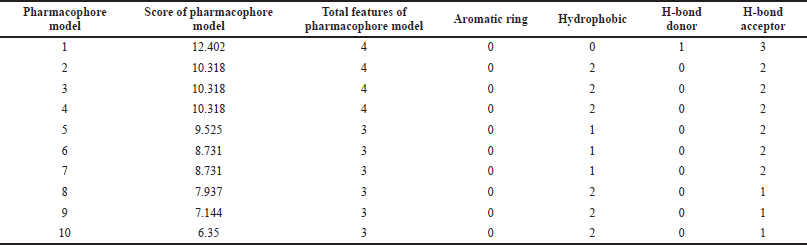 | Table 2. Detailed information on generated pharmacophore models from PharmaGist tool. [Click here to view] |
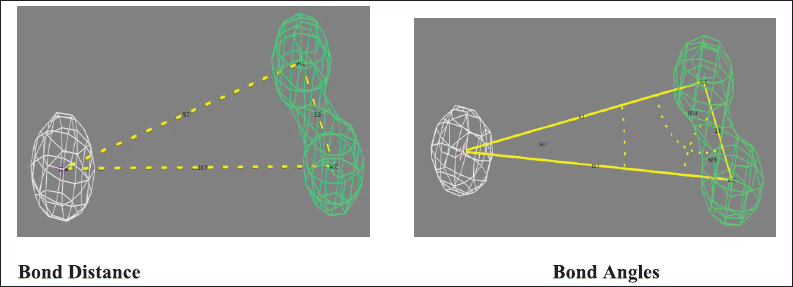 | Figure 4. Illustrating the bond distances and bond angles of selected pharmacophore model. [Click here to view] |
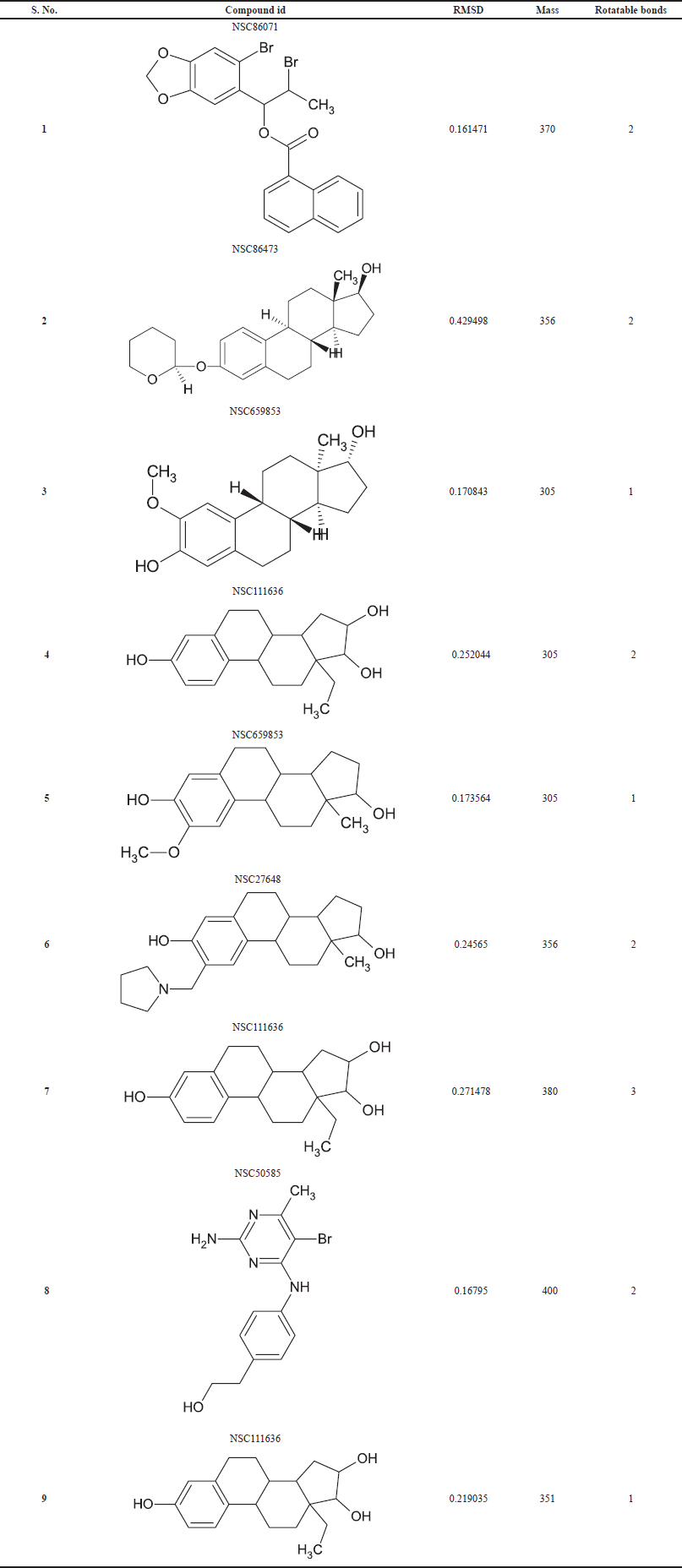 | Table 3. Molecular hits details using the pharmacophore model. [Click here to view] |
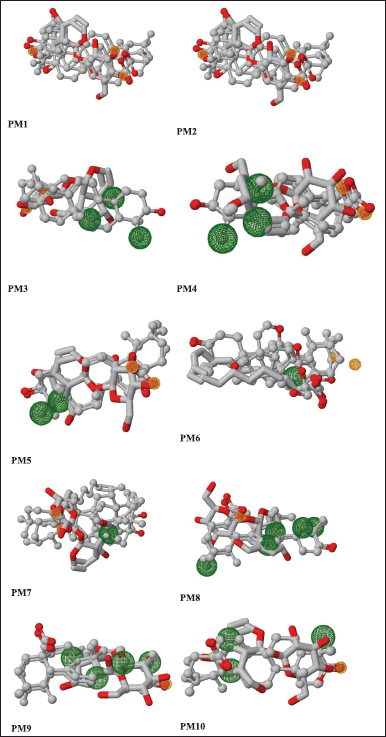 | Figure 5. Pairwise alignment of ursolic acid and selected ligands. Pharmacophore models (PM1–PM10) are represented as ball and stick models. [Click here to view] |
For all these 14 molecules, the docking scores above nine were taken into consideration. During this exercise, the obtained nine hits having NCI IDs NSC86071, NSC86473, and NSC111636, have binding energy scores nearer and more than the standard molecule, i.e., Celecoxib (−9 Kcal/mol). Finally, NSC86473 (−10 Kcal/mol) and NSC111636 (−9.3 Kcal/mol), have binding energy scores more than a standard molecule. Consequently, NSC86473 can behave as a powerful inhibitor against Cyclooxygenase 2. The binding energy scores and interactions are shown in the following tables and figures.
Interaction studies of screened hits
A ligand interaction diagram was created with discovery studio to analyze the interactions of hits attained from screening as shown in Figure 6. Hydrogen bond interactions were highlighted with green-colored lines. The ligand interactions for a target protein with top energy score hits are illustrated in Figure 6. The standard ligand (Celecoxib) shows five hydrogen bond interactions with amino acids TYR:373, GLN:370, TYR:122, ILE:124, ASP:125 with binding energy −9 Kcal/mol and five hydrophobic interactions with amino acid residues PHE:367, LEU:366, GLN:369, PHE:371. Among the hits, NSC86473 gives more binding interactions and energy than the standard ligand. It exhibited one polar hydrogen bond interaction with GLN:370, one polar hydrogen bond interaction with ARG:44, one polar hydrogen bond interaction with PHE:371, and two hydrophobic interactions with GLN:369, PRO:542. NSC111636 shows one polar hydrogen bond interaction with LYS:532 and three hydrophobic interactions with TYR:373, PRO:542, and LEU:366. NSC86071 shows one polar hydrogen bond interaction with GLN: 372 and two hydrophobic interactions with ARG: 44 and PRO: 542. NSC50585 shows four polar hydrogen bond interactions with GLU:465, CYS:36, ALA:156, and CYS:37 and four hydrophobic interactions with VAL:155, ASN:39, PRO:153, and PRO:40.
As per our results, we have identified the lead molecule that has a higher docking score than the standard Celecoxib and was found to satisfy drug-likeliness properties. We are planning to extend our work further by performing animal studies with ethical approval in our future projects.
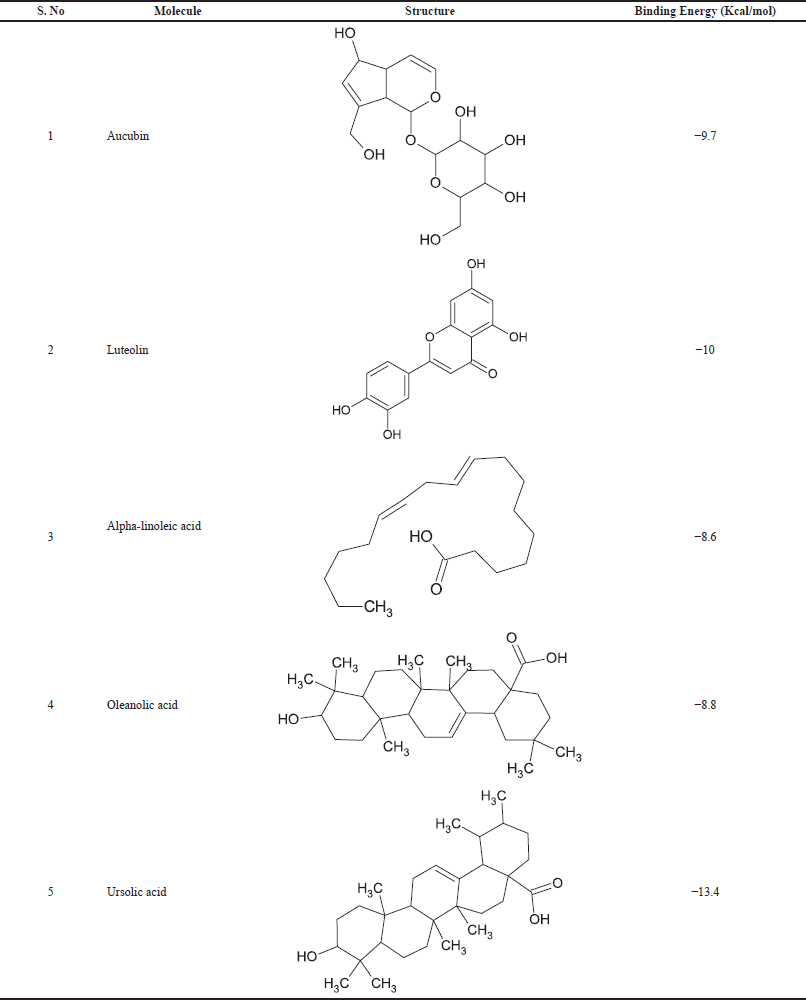 | Table 4. Selected phytoconstituents for pharmacophore generation and the binding energy values with protein (PDB ID:4COX) after docking. [Click here to view] |
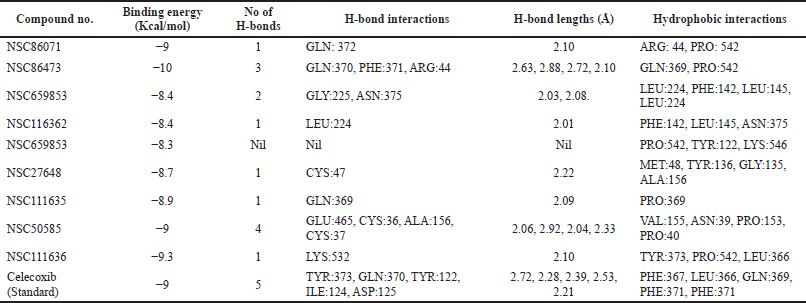 | Table 5. Molecular docking interactions, binding energy values of hits retrieved from Pharmit (NCI Database) with protein (PDB ID:4COX). [Click here to view] |
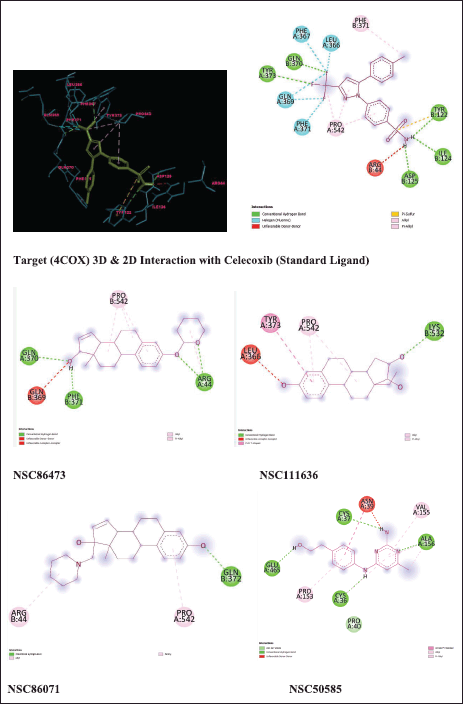 | Figure 6. Ligand interactions with target 4COX. [Click here to view] |
CONCLUSION
This study was designed to identify the herbs that are active in treating various diseases and is dynamic for future disease control programs. To attain this purpose, different computational tools like pharmacophore-based virtual screening, molecular docking, and binding free energy analysis were employed. A four-point pharmacophore hypothesis was recognized using five phytoconstituents of plant P. major and developed a PM1 in which all the input ligands were properly aligned and were feasible to screen the NCI database. The screened compounds were docked into the active site of 4COX and further Absorption, Distribution, Metabolism, Excretion properties were calculated. The results displayed that the compound NSC86473 may act as a powerful inhibitor against Cyclooxygenase 2 as it has the lowest binding energy than the standard and also had specified pharmacophoric features according to the developed PM1 model. In accordance with earlier findings, it can provide a few insights to research scholars in the future to identify and design new lead molecules with effective anti-inflammatory activity.
ACKNOWLEDGMENT
The authors are thankful to the Sarojini Naidu Vanita Pharmacy Maha Vidyalaya, Department of Pharmaceutical Chemistry for providing the necessary facilities and support to carry out this work.
AUTHORS CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
FUNDING
There is no funding to report.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in thçis research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Adom MB, Taher M, Mutalabisin MF, Amri MS, Abdul Kudos MB, Sulaiman MWAW, Sengupta P, Susanti D. Chemical constituents and medical benefits of Plantago major. Biomed Pharmacother, 2017; 96:348–60. CrossRef
Azab A, Nassar A, Azab AN. Anti-inflammatory activity of natural products. Molecules, 2016; 21(10):1321. CrossRef
Dror O, Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Novel approach for efficient pharmacophore-based virtual screening: method and applications. J Chem Inf Model, 2009; 49:2333–43. CrossRef
Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and innate immunity in infections and chronic inflammation. J. Autoimmun, 2015; 60:1–11. CrossRef
Kaserer T, Beck KR, Akram M, Odermatt A, Schuster D. Pharmacophore models and pharmacophore-based virtual screening: concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules, 2015; 20(12):22799–832. CrossRef
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH. PubChem substance and compound databases. Nucleic Acids Res, 2016; 44(Database issue):D1202–13. CrossRef
Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature, 1996; 384:644–8. CrossRef
Langer T, Hoffmann RD. Virtual screening an effective tool for lead structure discovery. Curr Pharm Des, 2001; 7(7):509–27. CrossRef
Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery principles, applications and recent advances. Curr Top Med Chem, 2014; 14(16):1923–1938. CrossRef
Deng W, Du H, Liu D, Ma ZC. Natural anti-inflammatory agents for pain relief. 2010; 1:80. CrossRef
Mozaffarian V. Identification of medicinal and aromatic plants of Iran. Farhang Moaser, Tehran, Iran, 2012.
MuniSireesha S, Dharani A, Donna Kanthi B, Sushma B, Pallavi C, Jhansi C, Saritha Jyostna T. Targeting the 3bgq - Pim1 kinase interaction with a series of novel dithiocarbamate substituted 2- oxindole derivatives - in silico studies. J Fac Pharm Ankara Ankara Ecz Fak Derg, 2022; 46(1):86–102. CrossRef
Muni Sireesha S, Dipankar Bhowmik, Soujanya D, Brijitha G, Jyothi V. Computational validation of tacrine analogs as antialzheimer’s agents against acetylcholinesterases. Int J Biol Pharm Allied Sci, 2021; 10(10):243–54. CrossRef
Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod, 2007; 70(3):461–477. CrossRef
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform, 2011; 3:33. CrossRef
Saikiran Reddy P, Sree Kanth S, Vijjulatha M. Discovery and design of new PI3K inhibitors through pharmacophore-based virtual screening, molecular docking, and binding free energy analysis. Struct Chem, 2018; 29:1753–66. CrossRef
Samuelsen AB. The traditional uses, chemical constituents and biological activities of Plantago major L. a review. J Ethnopharmacol, 2000; 71(1–2):1–21. CrossRef
Schneidman-Duhovny D, Dror O, Inbar Y, Nussinov R, Wolfson HJ. PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res, 2008; 36(Web Server issue):W223–8. CrossRef
Sunseri J, Koes DR. Pharmit: interactive exploration of chemical space. Nucleic Acids Res, 2016; 44(Web Server issue):W442–8. CrossRef
Usha S. Pharmacophore-based database searching of kinase-inhibitor mimetic molecular hits. J Bio Innov, 2016; 5:446–63.
Wang H, Zhao C, Huang Y, Wang F, Li Y, Chung HY. Chemical constituents and bioactivities of Plantaginis Herbs. Hong Kong Med J, 2015; 22:29–35.