
INTRODUCTION
Amikacin is a broad-spectrum, semi-synthetic amino-glycoside antibiotic that inhibits protein synthesis in a concentration-dependent manner by irreversibly binding to the bacterial 30s ribosome (Fourmy et al., 1996; Illamola et al., 2016; Krivoy et al., 1998; Mingeot-Leclercq et al., 1999). In the biopharmaceutical classification system, amikacin is placed in class III due to its poor oral bioavailability and permeability (Fatima et al., 2019). As a result, it is administered intravenously or intramuscularly and is predominantly excreted by the kidneys (Vogelstein et al., 1977). It is an antibiotic that is extensively used in newborn critical care units for meningitis, pneumonia, intra-abdominal infections, sepsis, joint infections, and urinary tract infections (Logre et al., 2020). Amikacin shows significant variations in pharmacokinetic (PK) behavior in neonates due to their highly dynamic physiological state. Sufficient PK studies need to be conducted in this vulnerable population to establish proper dosage regimens (Cristea et al., 2017). Additionally, therapeutic drug monitoring (TDM) needs to be conducted in patients with comorbidities and in conditions such as hypothermia and kidney failure to ensure that the treatment does not cause toxicity (Bleyzac et al., 2001; Saez Fernandez et al., 2019). There is an imminent need for a cost-effective, sensitive, reproducible, and reliable bioanalytical technique (Bleyzac et al., 2001; De Cock et al., 2012; Gijsen et al., 2021; Illamola et al., 2016; Klein et al., 1992; Wargo and Edwards, 2014).
Many analytical techniques have been reported for the quantitative analysis of amikacin (Bleske et al., 1987; Glinka et al., 2020; Stead, 2000; Thompson and Burd, 1980). However, the specificity, sensitivity, and adaptability of these methods are constrained by the chemistry of amikacin. Immunoassays have limitations due to the cross-reactivity of concurrently administered medications, metabolites, and endogenous compounds from complex matrices (Mehta, 1992; Tate and Ward, 2004). The chromatographic analysis of amikacin is complicated by its retention and detection issues. Due to the lack of a chromophore in the amikacin structure (Fig. 1), the high-performance liquid chromatography-UV-visible spectroscopy (HPLC-UV) based methods need derivatization, which raises the bar for accuracy and reproducibility. Amikacin molecules have multiple ionic groups (four amine and eight hydroxy groups), which have a major influence on retention and ionization patterns and cause difficulties in retention in a normal C18 column and require complex procedures. This intricate procedure becomes practically challenging when applied to quantitative analysis. The problem with poor chromatographic column retention could be resolved using ion-pairing reagents such as pentafluoropropionic acid (PFPA) and heptafluorobutyric acid (HFBA) anhydride (da Silva et al., 2020; Dijkstra et al., 2014). However, the use of ion-pairing reagents is constrained by ion suppression in LC-MS, prolonged equilibration times, lower column life, and the need for frequent ion source cleaning (Annesley, 2003; Gustavsson et al., 2001). A few chromatographic separations using ammonia solution with a high pH have been reported without the use of ion-pairing agents or derivatization (Chan et al., 2020), but the extreme column operating conditions restrict its routine use. Hydrophilic interaction liquid chromatography (HILIC) based separation is a good research option to overcome these challenges (Oertel et al., 2004).
HILIC columns consist of a wide variety of silica gel and polymer surfaces, chemically modified with diol, amine, amide, cyno, and carboxyl groups, and retain polar analytes by hydrogen bond or ionic interaction. For analytes with polycationic functional groups that make them very polar, like aminoglycoside antibiotics (amikacin), the best way to separate them is with a mixed-mode HILIC ion-exchange column with a polar surface and a modified carboxyl group (Buszewski and Noga, 2012).
To get around the problems and unrepeatable results of earlier methods that used derivatization and ion pairing reagents, this work aimed to develop and test a HILIC LC-MS/MS-based analytical technique for figuring out how much amikacin is in neonatal plasma samples.
MATERIALS AND METHODS
Reference standards, reagents, and biological sample
Tobramycin (internal standard) and amikacin sulfate were supplied by Sigma Aldrich (Sigma-Aldrich, St. Louis, MO) as secondary pharmaceutical standards. Strong ammonia solution and LC-MS grade acetonitrile were provided by Merck, India. Merck Millipore Direct Q-3 UV water purification system was used to obtain Type-I water. Clinical samples were collected after informed consent from patients meeting the inclusion criteria. The Institutional Ethical Committee (IEC) approval was obtained from Kasturba Medical College and Kasturba Hospital Institutional Ethics Committee (Registration No. ECR/146/Inst/KA/2013/RR19), Manipal dated August 13, 2019 (certificate number 558/209) and Clinical Trials Registry, India dated October 22, 2019 (CTRI/2019/10/021750), respectively. Whole blood and plasma samples from healthy adult human volunteers were used for method development and validation.
Apparatus
The analysis was performed on Dionex Ultimate 3000 HPLC from Thermo Fisher Scientific LLC. It was equipped with a temperature-controlled autosampler and column compartment connected to an LTQ-XL mass spectrometer. A polymer-based HILICpak VC-50 2D (150 × 2.1) mm, 5 μ column from Shodex, Japan, was used for the analysis. Ryan Labs, an Indian company, provided autosampler vial inserts with a 0.3 ml volume. The K2 EDTA tubes used to collect blood samples were purchased from CML Biotech Private Limited, India. The Sanyo Ultra-Low freezer, China was used for the storage of samples to study the long-term stability. The short-term stability samples, working standard solutions, and standard stock solutions were all stored at 2°C–8°C in a Godrej Eon refrigerator, India. During various stages of sample preparation, the samples were centrifuged in a REMI C24 cooling centrifuge that had a rotor head of R-248 M (24 × 1.5 ml).
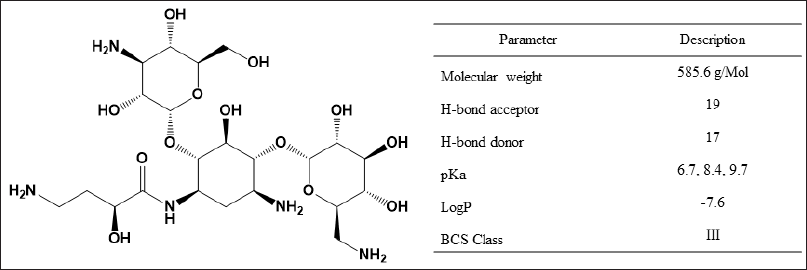 | Figure 1. Structure and chemistry of amikacin. [Click here to view] |
Chromatography conditions
Gradient programming of mobile phase A (1.5% v/v ammonia solution in water) and mobile phase B (acetonitrile) at a flow rate of 0.2 ml/minute and a column oven temperature of 40°C has been used to obtain the chromatographic separation. The gradient program started with an initial 70% A and increased linearly to 90% A over 5 minutes. It was then held for 5 minutes before returning to its initial condition in 1.5 minutes and kept for conditioning for 4.5 minutes. The injection volume was 5 μl, and the autosampler temperature was maintained at 8°C.
Mass spectrometry
The mass spectra ionization source was heated electrospray ionization that was run under the following conditions: spray voltage (4.20 kV), heater temperature (380°C), capillary voltage (45 V), capillary temperature (350°C), tube lens offset voltage (140 V), sheath gas flow rate (45 arb), auxiliary gas flow rate (12 arb), and sweep gas flow rate (0 arb) units. The collision-induced dissociation (CID) for tobramycin m/z 586.0→424.6 was achieved at a normalized collision energy of 18.0% with an isolation width of 4.0 and the CID of amikacin m/z 468.0→325.0 was achieved at a normalized collision energy of 35.0% with an isolation width of 4.0, respectively in positive MS/MS scan.
Preparation of stock solution
Amikacin and tobramycin standard stock solutions in water were prepared at concentrations of 6,813.0 and 1,000.0 μg/ml, respectively. Working standards of amikacin in the range of 12.6–2,500.0 μg/ml were prepared by serial dilution of stock. A working standard of internal standard (IS) having a concentration of 300.0 μg/ml was prepared from tobramycin stock solution in water.
Calibration and quality control (QC) samples
Healthy adult human pooled plasma was spiked at 4% (v/v) with the appropriate working standard solution to get the calibrator concentrations of 0.5, 1, 2, 4, 8, 25, 50, 65, 80, and 100 μg/ml. QC samples were generated in the same manner, with concentrations of 0.5, 1.5, 40, and 80 μg/ml, indicating the lower limit of quantification (LLOQ), lower QC (LQC), medium QC (MQC), and higher QC (HQC).
Clinical sample collection
A prospective longitudinal observational study was designed to collect plasma samples from patients for clinical validation. The neonates who met the inclusion criteria for this study and received amikacin as part of their routine treatment were enrolled after obtaining the necessary consent and IEC clearance. Certified nurses collected the blood samples, which were centrifuged at 4,600× g for 5 minutes to obtain a clear plasma specimen and stored at −80°C until analysis.
Plasma sample preparation
Into a 1.5-ml conical-shaped centrifuge tube, 10 μl of working standard solution of IS and 50 μl of plasma sample were taken and mixed. The above mixture was precipitated with 160 μl of cold acetonitrile, extracted with 160 μl of water, and centrifuged at 9,392× g at 4°C for 10 minutes to get a clean supernatant for LC-MS/MS analysis.
Method validation
The analytical method was validated as per the US-FDA and ICH M10 guidelines (ICH, 2022; U. S. Food and Drug Administration, 2018). The evaluation included parameters like system suitability, selectivity, carry-over, matrix effect, recovery, linearity, accuracy, and precision as well as dilution integrity and stability.
System suitability
A set of six MQC replicate samples were analyzed to ensure the system’s performance. This is evaluated by monitoring the column back pressure, retention time, and area response.
Selectivity and carry-over
A selectivity study was performed to determine the ability of the method to differentiate and measure any potential interference present in the blank matrix. Six blank plasma matrices from different individuals were sourced and injected after LLOQ. There should not be any interference at the retention time of the analyte.
The difference in the measured concentration of analyte/IS caused by residual analyte/IS from the prior sample that remains in the autosampler injector was also evaluated by injecting a blank matrix after the LLOQ and upper limit of quantification (ULOQ) in six replicates. Responses in the blank matrix at analyte retention time should be less than 20% of LLOQ, and less than 5% of LLOQ for IS to ensure that there is no carry-over.
Matrix effect
Amikacin was spiked at LQC and HQC in three replicates from six healthy individuals to study the effects of different plasma sources on amikacin measurement and evaluated to ensure accuracy within ±15% of the nominal concentration and precision % Coefficient of variance (CV) not more than 15%.
Linearity and LLOQ
Three independent calibration curves (CC) were performed on three different days in the range of 0.5–100 μg/ml calibrators. The linearity curves were plotted by taking weighing factor 1/X in Qual browser, XCalibur software, and calculating the slope and intercept. The back-calculated concentration of calibrator standards was assessed to be within ±20% of the nominal concentration of amikacin for LLOQ and ±15% for other CC standards, indicating that the approach is appropriate for the tested range. The LLOQ was set by the visual method by observing the precision from six replicate injections and a signal-to-noise ratio not less than 10 at 0.5 μg/ml.
Accuracy and precision
Six replicates of QC samples were injected at LLOQ, LQC, MQC, and HQC to assess accuracy and precision within the run. Three separate runs for accuracy and precision over successive days were used to establish between-run accuracy and precision. An acceptable limit of ±15% accuracy (±20% for LLOQ) and ≤15% CV precision was used to evaluate the bias of computed concentrations using the nominal concentrations of QCs and the coefficient of variation. Prior to each accuracy and precision run, a linearity run was performed for compliance with the acceptance limit as indicated in the linearity range.
Recovery
The pre-extraction and post-extraction spiked QC and IS working standard was performed in five replicates at LQC and HQC to assess the extent and uniformity of analyte and IS extraction from plasma by direct area-to-area comparison method with an acceptance limit of ≤10% coefficient of variation from mean recovery.
Dilution integrity
A 2.5 × ULOQ (250 μg/ml) of amikacin from an independent stock solution was spiked into the blank plasma and analyzed after diluting 3–4 times with blank plasma to ensure the dilution integrity of this method. The back-calculated concentrations of the integrity samples should be within the tolerance level of ±15% accuracy and precision %CV not more than 15%.
Stability
The stability of the standard stock solution of amikacin and internal standard was assessed at 2°C–8°C on 0, 3, 7, 15, and 30-day time points. The stability of amikacin in plasma matrix at room temperature (bench-top stability, 24 hours), autosampler stability (at 8°C, 24 hours), processed sample stability (at 2°C–8°C freezer, 48 hours), freeze-thaw stability (at −80°C, five cycles at 24 hours interval) were evaluated at low concentration and high concentration QCs. The long-term stability of amikacin in the plasma matrix at −80°C was evaluated on 0, 3, 7, 15, 30, and the 60-day time points both at LQC and HQC.
Clinical validation
The clinical study was carried out with the approval of the institutional ethics committee. A total of 45 samples were collected from 33 neonates at random times from subjects undergoing routine clinical investigations and analyzed in 2 separate sequences with independent calibration standards. The study findings were compared to the US-FDA recommendation of a peak in the range of 24–35 μg/ml and a trough in the range of 2–5 μg/ml for patients receiving amikacin (Cristea et al., 2017). Peak is defined as 1 hour after the start of the infusion or 30 minutes after the end of the infusion, and trough is defined as within 30 minutes of giving the next dose.
RESULTS AND DISCUSSION
Method optimization
During method development, we considered all the possible sources of errors with respect to the analysis, as shown in Figure 2 (Annesley, 2003; Sonawane et al., 2019). Due to its strong polarity, amikacin is not retained in many reverse-phase columns used in chromatography. This presented a substantial obstacle to overcome while developing a chromatographic procedure for amikacin. As shown in Table 1, conventional methods have either employed high-pH aqueous ammonia solution or reverse-phase columns with volatile ion-pairing reagents in the mobile phase. Ion pairing reagents, however, lead to ion suppression in mass spectrometers, making them unfit for routine analysis.
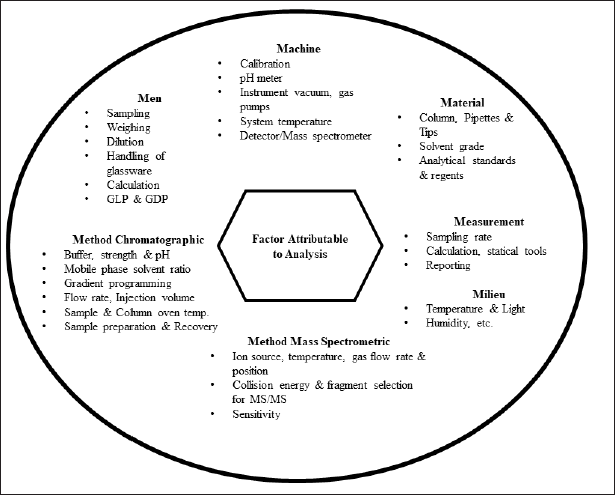 | Figure 2. Factors attributable to analytical method development and analysis in LC-MS/MS. [Click here to view] |
Due to the polar nature of aminoglycosides, we investigated the suitability of a mixed-mode hydrophilic cation-exchange HILICpak VC-50 2D column as an alternative to the more often employed reverse-phase columns. HILIC ion exchange columns can withstand high pH of up to 12, which is an essential requirement for the chromatographic separation of amikacin. The HILICpak VC-50 2D column is recommended for use with a flow rate of 0.1–0.3 ml/minute, an oven temperature of 4°C–60°C, and a maximum back-pressure limit of 100 bars. A good peak shape, retention time, and mass spectrometer sensitivity were achieved by maintaining a flow rate of 0.2 ml/minute and a column oven temperature of 40°C, respectively, at a back-pressure of around 84 bars. The ideal condition for HILIC chromatography is to use a higher ratio of organic solvent compared with the aqueous phase to retain the polar analytes. However, in the case of the HILICpak VC-50 2D (150 × 2.0) mm column, the eluent required a higher ratio of strong basic pH in the aqueous phase to reduce the ionic strength. Therefore, the polarity of the mobile phase was increased by increasing the aqueous ammonia solution over the organic phase (acetonitrile) from 10% to 90% as a trial-and-error method. Reducing the run time and obtaining a sharp peak shape in the HILICpak column was another challenge compared to the reverse phase column. The final gradient program was set as initially 30% acetonitrile, brought linearly to 10% in 5 minutes, and kept constant at 10% acetonitrile for another 5 minutes for elution of both internal standards and amikacin. The use of lower pH conditions led to the ionization of amikacin, causing a strong ionic interaction with the stationary phase leading to tailing and a broad peak shape. Using an alkaline pH of 11.5 combined with 1.5% ammonia produced the best peak shape and elution results. Since the pKa value of amikacin is about 9.7, it is unionized at a pH of 11.5. This reduces the ionic interaction between the analyte and the cationic functional group of the stationary phase, resulting in a sharp peak (Kulkarni et al., 2016a, 2016b). In this method, linearity in the range of 0.5–100 μg/ml was chosen for TDM in neonates, considering that the Cmax of amikacin is below 60 μg/ml according to published literature (Cristea et al., 2017; Illamola et al., 2016; Smits et al., 2015). The method was set to a 5-μl injection volume based on the area response of amikacin at LLOQ.
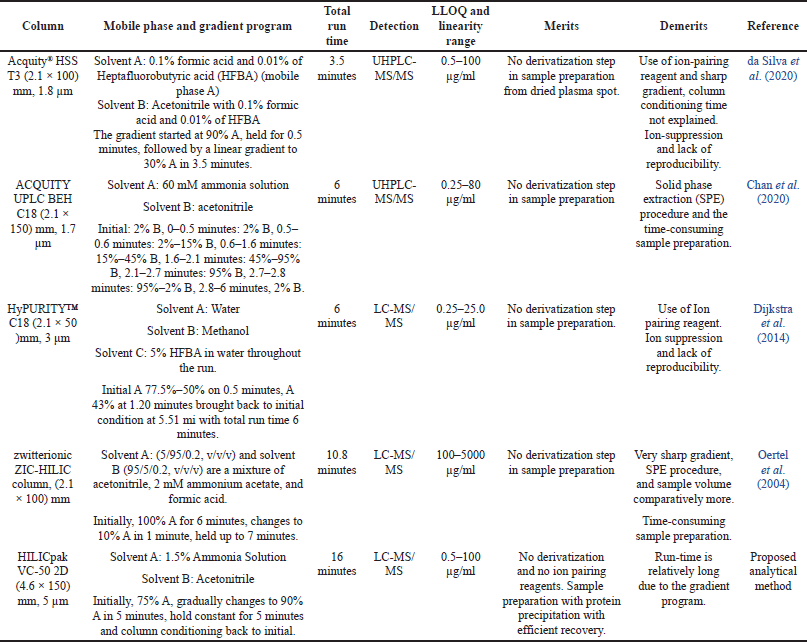 | Table 1. The previously reported bioanalytical LC-MS/MS method for amikacin. [Click here to view] |
System suitability
The system suitability test had IS and amikacin retention time within 3.0 ± 0.30 and 10.0 ± 1.0 minutes, respectively, with column back-pressure fluctuation of not more than three bars. Due to the polycationic nature of the analytes, it was observed that even a slight change in the composition of the mobile phase had a significant impact on the MS response. Therefore, the strength and pH stability of the 1.5% (v/v) ammonia solution and the MS scan event were deemed to be of utmost importance. The representative chromatograms of LLOQ at 0.5 μg/ml, ULOQ at 100 μg/ml, and the mass spectrum are depicted in Figure 3.
Selectivity and carry-over
The responses in the blank plasma samples at the retention time of amikacin and IS were less than 20% and 5%, respectively, compared to the plasma-spiked LLOQ for the selectivity and carry-over run.
Matric effect
To study the matrix effect of plasma, spiked plasma LQC and HQC were prepared in three replicates from the plasma of six different healthy individuals. The overall accuracy ranged from 95.22% to 96.60% with a coefficient of variance of 3.18% to 5.30% at LQC and 94.46% to 98.44% with a coefficient of variance of 2.45% to 5.72% at HQC, demonstrating an acceptable matrix effect of ≤15% of the nominal concentration and precision of ≤15% CV.
Linearity range
Each of the independent CC runs between 0.5 and 100 μg/ml produced a regression coefficient (r²) of not less than 0.99, showing a strong correlation between the area ratio of the analyte to the internal standard. Results are summarized in Table 2. The back-calculated concentration of calibrator standards was within ±20% of the nominal concentration of amikacin for LLOQ and ±15% for other CC standards, indicating that the method is appropriate for the tested range.
Accuracy and precision
Prior to each accuracy and precision run, a linearity run was assessed for compliance with the acceptance limit indicated in the linearity range. The back-calculated concentrations of spiked plasma QCs were within acceptable limits of accuracy and precision, as shown in Table 3. Within-run accuracy in three different runs was between 93.61% and 97.75%, and the precision was between 2.45% and 6.06%. Between-run accuracy from three separate runs was 94.70%–98.10%, with a precision of 3.82%–5.47%.
Recovery
Amikacin recovery from pre-extraction compared to the post-extraction spiked QC plasma samples, showed 97.32% assay (5.15% CV) at LQC and 98.39% assay (3.34% CV) at HQC for amikacin and for IS (tobramycin) was 98.65% (4.51% CV).
Dilution integrity
Back-calculated concentrations of the three- and four-times dilutions of the integrity sample showed 96.27% and 97.01% assay with coefficients of variation of 2.97% and 3.50%, respectively. These results remained within the tolerance level of ±15% accuracy and <15% precision from nominal concentrations.
Stability evaluation
The internal standard and stock solution of amikacin exhibited stability over 30 days, with a total deterioration of 2.1% ± 1.8% and 3.2% ± 1.5%, respectively, from the initial concentration. Table 4 lists the results of experiments on the stability of the autosampler, bench-top, freeze-thaw, and processed samples at LQC and HQC levels, all of which revealed satisfactory stability. Figure 4 illustrates the long-term stability of amikacin in plasma stored at −80°C. The 60th day of plasma stability samples revealed a 6.56% ± 1.8% decrease from the initial concentration, indicating no significant reduction in amikacin concentration.
Clinical validation
All term-neonates enrolled in this study received antibiotic therapy as part of a hospital standard care plan based on their body weight, post-natal age, gestational age, and sepsis consideration, among other factors. The demographic details are provided in Table 5. A sparse sampling approach was utilized to avoid sampling intervention. All collected samples’ plasma amikacin concentrations were analyzed using the developed method and clinically correlated for the observed levels. Amikacin plasma concentrations were expected to range between 2 and 35 mg/l for the minimum effective concentration (MEC) and maximum safe concentration (MSC), respectively. Figure 5 shows the time after dosing versus plasma concentration for all samples collected. Few plasma concentrations were found to be outside the therapeutic range (2–35 mg/l). Three neonates with serum amikacin concentrations greater than 35 mg/l had serum creatinine levels greater than 0.5 mg/l, indicating low plasma clearance, which could be due to underdeveloped kidneys or deteriorated kidney function. Subjects with serum creatinine levels greater than 0.7 mg/dl had plasma concentrations of amikacin greater than 3 mg/l even after 30 hours of dosing, indicating that lower clearance of amikacin correlates with higher serum creatinine levels due to underdeveloped or deteriorated kidney function. Meanwhile, all other neonates with serum creatinine levels less than 0.3 mg/dl achieved plasma amikacin concentrations within the therapeutic window and fell below 2 mg/l within 24 hours, as expected. These findings are clinically correlating with the observed concentration in our assay.
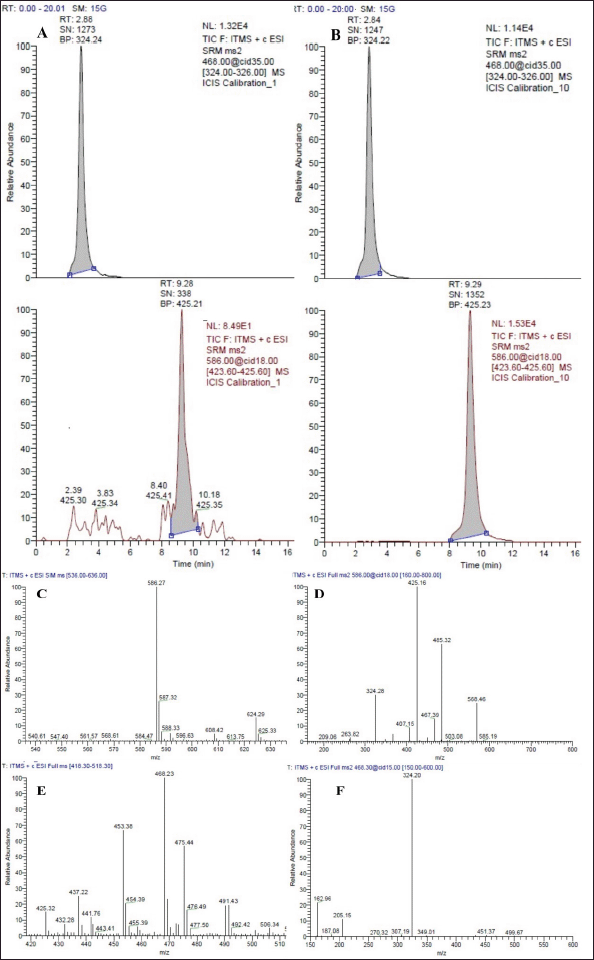 | Figure 3. (A) Chromatogram of amikacin (RT 9.26 minutes) and internal standard (RT 2.88 minutes) (Tobramycin) at LLOQ. (B) Chromatogram of amikacin (RT 9.29 minutes) and internal standard (RT 2.84 minutes) (Tobramycin) at ULOQ. (C) Mass spectrum showing Amikacin precursor ion [M + H]+, 586.27 m/z. (D) Mass spectrum showing amikacin fragment ion (Top m/z peak [M + H]+, 586.27→425.16 m/z). (E) Mass spectrum showing tobramycin precursor ion [M + H]+, 468.23 m/z. (F) Mass spectrum showing tobramycin fragment ion (Top m/z peak [M + H]+, 468.23→324.20 m/z). [Click here to view] |
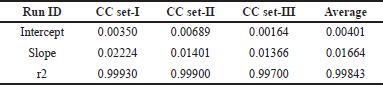 | Table 2. Intercept, slope, and correlation coefficient of three CC plots. [Click here to view] |
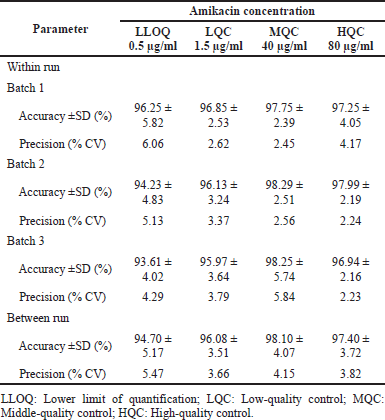 | Table 3. Accuracy and precision data of QC samples of amikacin from spiked plasma. [Click here to view] |
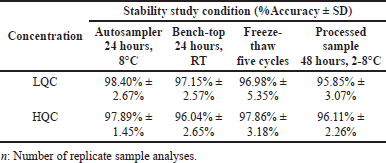 | Table 4. Amikacin stability results under different conditions. study. [Click here to view] |
Greenness of analytical procedure
In order to evaluate the eco-friendliness of the proposed analytical method, a total of 12 GAPI parameters pertaining to the sample, the reagents and compounds, and the instruments were analyzed. Collecting, preserving, transporting, storing, processing methods, extraction scale, solvent or reagent employed, and additional reagents are sample variables. The amount of solvent/reagent, health hazards, and safety hazards were associated with chemicals and reagents. Instrumentation was related to energy, occupational danger, waste, and waste treatment (P?otka-Wasylka, 2018). A pictogram (Fig. 6) was constructed using the GAPI tool, which indicates that the method is eco-friendly. A circle in the middle of a pictogram represents that the method is suitable for both quantitative and qualitative purposes. The color red represents a severe danger to the environment, while yellow and green symbolize lower danger and better greenness. Overall, the method was eco-friendly, with five green, seven yellow, and three red.
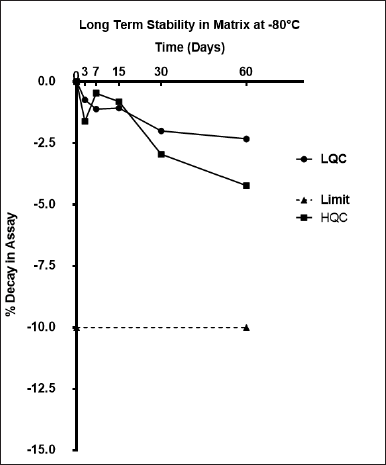 | Figure 4. Long-term stability data of amikacin in plasma at −80°C. [Click here to view] |
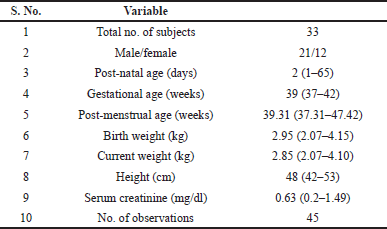 | Table 5. Summary of demographic details of neonatal patients. [Click here to view] |
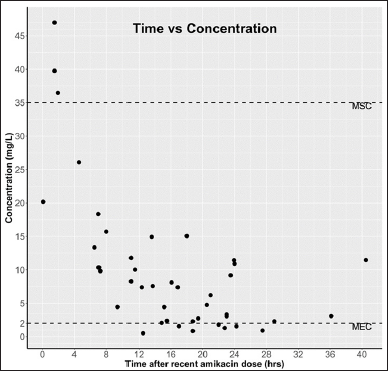 | Figure 5. Time versus concentration of amikacin measured in 45 plasma samples of neonates (MSE: maximum safe concentration and MEC: minimum effective concentration). [Click here to view] |
 | Figure 6. Green Analytical Procedure Index pictogram of proposed LC-MS/MS method for estimation of amikacin from neonatal plasma. The color red represents a high danger to the environment, while yellow and green symbolize lower danger and better greenness. Eight parameters relating to sample (collection, preservation, transport, storage, types of processing method, scale of extraction, solvent/reagent used and additional reagent, etc.), three parameters relating to reagents and compounds (amount to solvent/reagent, health hazard, and safety hazard) and four parameters relating to instrumentation (energy, occupational hazard, waste, and waste treatment). [Click here to view] |
CONCLUSION
A simple, efficient, reproducible, and green analytical method was developed and validated for the estimation of amikacin levels in neonatal plasma for PK evaluations and TDM. The method does not use any derivatization steps or any ion-pairing reagents. Clinical validation of the method demonstrated that the observed concentrations of amikacin correlate with the clinical conditions present in the neonatal subjects. The concentration of amikacin was found to fall outside the predicted therapeutic window for clinically unstable neonates in the usual dosing regimen. The developed technique could be used for neonatal population PK investigations and could be adapted to clinical laboratories for neonatal therapeutic monitoring of amikacin. The method will also find its usefulness in bioavailability and bioequivalence studies of amikacin.
ACKNOWLEDGMENTS
We thank the Indian Council of Medical Research for funding this research project under the Extramural Ad-hoc grant and the Manipal Academy of Higher Education for providing the research facilities. We appreciate the technical advice provided by SHOWA DENKO K.K., Japan, during the analysis.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study was approved from the Institutional Ethical Committee (IEC), Kasturba Medical College and Kasturba Hospital Institutional Ethics Committee (Registration No. ECR/146/Inst/ KA/2013/RR19), Manipal dated August 13, 2019 (certificate number 558/209) and Clinical Trials Registry, India dated October 22, 2019 (CTRI/2019/10/021750), respectively.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Annesley TM. Ion suppression in mass spectrometry. Clin Chem, 2003; 49(7):1041–4. CrossRef
Bleske BE, Larson TA, Rotschafer JC. Observed differences in amikacin pharmacokinetic parameters and dosage recommendations determined by enzyme immunoassay and fluorescence polarization immunoassay. Ther Drug Monit, 1987; 9(1):48–52. CrossRef
Bleyzac N, Varnier V, Labaune JM, Corvaisier S, Maire P, Jelliffe RW, Putet G, Aulagner G. Population pharmacokinetics of amikacin at birth and interindividual variability in renal maturation. Eur J Clin Pharmacol, 2001; 57(6–7):499–504.
Buszewski B, Noga S. Hydrophilic interaction liquid chromatography (HILIC)—a powerful separation technique. Anal Bioanal Chem, 2012; 402(1):231–47. CrossRef
Chan K, Wang W, Ledesma KR, Yin T, Tam VH. A robust LC-MS/MS method for amikacin: application to cellular uptake and pharmacokinetic studies. Bioanalysis, 2020; 12(7):445–54. CrossRef
Cristea S, Smits A, Kulo A, Knibbe CAJ, van Weissenbruch M, Krekels EHJ, Allegaert K. Amikacin pharmacokinetics to optimize dosing in neonates with perinatal asphyxia treated with hypothermia. Antimicrob Agents Chemother, 2017; 61(12):e01282–17. CrossRef
da Silva ACC, de Lima FLL, Bastiani MF, Antunes MV, Brucker N, Linden R. Dried plasma spots for therapeutic monitoring of amikacin: validation of an UHPLC-MS/MS assay and pharmacokinetic application. J Pharm Biomed Anal, 2020; 184(1):113201. CrossRef
De Cock RF, Allegaert K, Schreuder MF, Sherwin CM, de Hoog M, van den Anker JN, Danhof M, Knibbe CA. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet, 2012; 51(2):105–17. CrossRef
Dijkstra JA, Sturkenboom MG, Hateren K, Koster RA, Greijdanus B, Alffenaar JW. Quantification of amikacin and kanamycin in serum using a simple and validated LC-MS/MS method. Bioanalysis, 2014; 6(16):2125–33. CrossRef
Fatima S, Panda AK, Talegaonkar S, Iqbal Z, Ahmad FJ. Optimization and designing of amikacin-loaded poly D, L-Lactide-co-glycolide nanoparticles for effective and sustained drug delivery. J Pharm Bioallied Sci, 2019; 11(1):83–95. CrossRef
Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science, 1996; 274(5291):1367–71. CrossRef
Gijsen M, Vlasselaers D, Spriet I, Allegaert K. Pharmacokinetics of antibiotics in pediatric intensive care: fostering variability to attain precision medicine. Antibiotics (Basel), 2021; 10(10):1182. CrossRef
Glinka M, Wojnowski W, Wasik A. Determination of aminoglycoside antibiotics: current status and future trends. Trends Anal Chem, 2020; 131(1):116034. CrossRef
Gustavsson SA, Samskog J, Markides KE, Langstrom B. Studies of signal suppression in liquid chromatography-electrospray ionization mass spectrometry using volatile ion-pairing reagents. J Chromatogr A, 2001; 937(1–2):41–7. CrossRef
Illamola SM, Colom H, van Hasselt JG. Evaluating renal function and age as predictors of amikacin clearance in neonates: model-based analysis and optimal dosing strategies. Br J Clin Pharmacol, 2016; 82(3):793–805. CrossRef
ICH. International Council for Hormonization of technical requirements for pharmaceuticals for human use, bioanalytical method validation, M10. ICH, 2022. Geneva, Switzerland.
Klein J, Koren G, MacLeod SM. Comparison of methods for prediction of nephrotoxicity during development. Dev Pharmacol Ther, 1992; 19(2–3):80–9. CrossRef
Krivoy N, Postovsky S, Elhasid R, Ben Arush MW. Pharmacokinetic analysis of amikacin twice and single daily dosage in immunocompromised pediatric patients. Infection, 1998; 26(6):396–8. CrossRef
Kulkarni BM, Bhamare VS, Santhakumari B. Oxidative transformation of antiretroviral drug zidovudine during water treatment with permanganate: reaction kinetics and pathways. Desalin Water Treat, 2016a; 57(52):24999–5010. CrossRef
Kulkarni RM, Bhamare VS, Santhakumari B. Mechanistic and spectroscopic investigations of Ru3+-catalyzed oxidative degradation of azidothymidine by heptavalent manganese at environmentally relevant pH, Desalin Water Treat, 2016b; 57(58):28349–62. CrossRef
Logre E, Enser M, Tanaka S, Dubert M, Claudinon A, Grall N, Mentec H, Montravers P, Pajot O. Amikacin pharmacokinetic/pharmacodynamic in intensive care unit: a prospective database. Ann Intensive Care, 2020; 10(1):75. CrossRef
Mehta AC. A critical appraisal of chromatographic and immunoassay techniques for clinical drug analysis. J Clin Pharm Ther, 1992; 17(6):325–31. CrossRef
Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother, 1999; 43(4):727–37. CrossRef
Oertel R, Neumeister V, Kirch W. Hydrophilic interaction chromatography combined with tandem-mass spectrometry to determine six aminoglycosides in serum. J Chromatogr A, 2004; 1058(1–2):197–201. CrossRef
P?otka-Wasylka J. A new tool for the evaluation of the analytical procedure: green analytical procedure index. Talanta, 2018; 181(1):204–9. CrossRef
Saez Fernandez EM, Perez-Blanco JS, Lanao JM, Calvo MV, Martin-Suarez A. Evaluation of renal function equations to predict amikacin clearance. Expert Rev Clin Pharmacol, 2019; 12(8):805–13. CrossRef
Smits A, De Cock RF, Allegaert K, Vanhaesebrouck S, Danhof M, Knibbe CA. Prospective evaluation of a model-based dosing regimen for amikacin in preterm and term neonates in clinical practice. Antimicrob Agents Chemother, 2015; 59(10):6344–51. CrossRef
Sonawane SS, Chhajed SS, Attar SS, Kshirsagar SJ. An approach to select linear regression model in bioanalytical method validation. J Anal Sci Technol, 2019; 10(1):1–7. CrossRef
Stead DA. Current methodologies for the analysis of aminoglycosides. J Chromatogr B Biomed Sci Appl, 2000; 747(1–2):69–93.
Tate J, Ward G. Interferences in immunoassay. Clin Biochem Rev, 2004; 25(2):105–20. CrossRef
Thompson SG, Burd JF. Substrate-labeled fluorescent immunoassay for amikacin in human serum. Antimicrob Agents Chemother, 1980; 18(2):264–8.
U. S. Food and Drug Administration. Bioanalytical method validation, guidance for industry. U. S. Food and Drug Administration, 2018. Maryland, United States of America. CrossRef
Vogelstein B, Kowarski A, Lietman PS. The pharmacokinetics of amikacin in children. J Pediatr, 1977; 91(2):333–9. CrossRef
Wargo KA, Edwards JD. Aminoglycoside-induced nephrotoxicity. J Pharm Pract, 2014; 27(6):573–7. CrossRef