
INTRODUCTION
The novel coronavirus (2019-nCoV) was first found in Wuhan, China, in December 2019 (Wu et al., 2020b; Xiantian et al., 2020). This novel coronavirus infection has been spreading globally and has become a pandemic. In February 2020, The International Committee on Taxonomy of Viruses classified 2019-nCov as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and the World Health Organization named the diseases caused by SARS-CoV-2 as COVID-19 (Gorbalenya et al., 2020; Wu et al., 2020a). The SARS-CoV-2 is a spherical-shaped virion with a positive-stranded RNA viral genome of size 30 kb translated into structural and nonstructural proteins (NSPs) (Guruprasad, 2021). RNA viruses, including SARS-CoV-2, have a high mutation rate up to a million times higher than their hosts (Pachetti et al., 2020). Coronavirus particles contain main structural proteins, including spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins, which also encode several nonstructural and accessory proteins (Arashkia et al., 2021). The S proteins are involved in binding host cellular receptors, angiotensin-converting enzyme 2, and subsequent membrane fusion. After translation, the polyproteins are cleaved by SARS-CoV-2 papain-like protease (PLpro) and 3-chymotrypsin-like cysteine protease (3CLpro, also called Mpro). The NSPs PLpro and 3CLpro are the SARS-CoV-2’s major important cysteine proteases, processing polyproteins translated from the viral RNA genome to produce the active functional proteins required for viral replication and, hence, for more infections (Zhang et al., 2020). The other essential enzyme in SARS-CoV-2 replication is helicase and RNA-dependent RNA polymerase (RdRp) complex for replication of the genome of positive-strand RNA. The S, M, E, and N proteins come together to form compartment endoplasmic reticulum and Golgi and then become a virion in the vesicle. The vesicle containing the virion penetrates the membrane plasma to release new viruses (Guo et al., 2020; Li et al., 2020; Maier et al., 2015). Due to the importance of these structural and NSPs of SARS-CoV-2 in the life cycle of the virus, inhibition of these proteins is expected to have a significant impact to disrupt the viral life cycle and obstruct the viral performance and rate of viral infection.
Natural products derived from plants, microorganisms, and animals have long been utilized to treat infectious and noninfectious disorders (Kim et al., 2016, 2018). A recent study reported the natural products which have antiviral activity and antioxidant activity could be a potential drug against the SARS-CoV-2 infection (Diniz et al., 2020; Islam et al., 2020). Brianthein V is a diterpene isolated from Briareum asbestinum and has cytotoxic and antiviral activities (Coval et al., 1988). Other diterpenes, such as ent-7alpha-hydroxykaur-16-en-19-oic acid, (-)-Kaur-16-en-7beta-ol-19-oic acid, and ent-7alpha-hydroxykaur-16-en-19-oate, are obtained from the methanolic extract of Plectranthus asirensis, a Yemeni folk medicinal plant used to cure disease and increase immunity (Saeed et al., 2020). It has been found that ent-11α-hydroxy15-oxo-kaur-16-en-19-oic-acid (also known as 5F), which is isolated from the herb Pteris semipinnata L. (Banbianqi in Chinese), can inhibit the growth of several tumor cell lines, including gastric adenocarcinoma cells and lung adenocarcinoma cells (Lu et al., 2013). Adenostemma lavenia (L.) O. Kuntze, also known as sticky daisy, grows widely in the tropical regions of Asia. Traditionally, this plant is used as a medicinal herb to cure pneumonia, lung congestion, fever, hepatitis, and skin wounds (Cheng et al., 1979). Previously, a study reported that ent-11α-hydroxy-15-oxo-kaur-16-en-19-oic acid from A. lavenia extracts as NRF2 activators can alleviate the symptoms of pneumonia in model animals (Batubara et al., 2020). It is also reported that p-coumaric acid-containing A. lavenia ameliorates acute lung injury by activating AMPK/Nrf2/HO-1 signaling and improving the antioxidant response (Chen et al., 2019). We expected these materials containing kaurene diterpenoids could be a potential preventive and therapeutic agent in the fight against the COVID-19 disease.
Computational screening, molecular docking, molecular dynamics (MD), and pharmacokinetics analysis are some of the fastest approaches to predict the potency of the compounds before in vitro and in vivo trials are performed and approved for clinical treatment of the patients. Therefore, this computational study was conducted to evaluate the effect of some kaurene diterpenoids and their derivatives on inhibiting SARS-CoV-2 nonstructural proteins (PLpro, 3CLpro) and structural proteins (membrane, nucleocapsid) and then compare them to medicines used for COVID-19 treatment, in the clinical phase, i.e., favipiravir and remdesivir.
MATERIALS AND METHODS
Materials and tools
In this study, we use the 3D structure of the receptor proteins of SARS-CoV-2, including NSPs (3CLpro (PDB ID: 6WNP) and PLpro (PDB ID: 6WX4)) and structural proteins (nucleocapsid (PDB ID: 6YVO) and membrane (QHD43419)). The structures were retrieved from the Protein Data Bank (PDB) database (http://www.rcsb.org/pdb/), except the membrane protein, which was obtained from the I-Tasser product by Zhang Laboratory Michigan University (https://zhanggroup.org//COVID-19/).
The structures of the ligands [20 kaurene diterpenoids and SARS-CoV-2 therapeutic drugs: favipiravir and remdesivir (Table 1)] were retrieved from the PubChem database (http://PubChem.ncbi.nlm.nih.gov) in the structure data file format (Kim et al., 2019). The software used includes AutoDock Tools (Morris et al., 2014), AutoDock Vina (Trott and Olson, 2010), Open Babel 3.0 (O’Boylen et al., 2011), VMD (Visual Molecular Dynamics) 1.9.3 (Humphrey et al., 1996), LigPlot+ (Wallace et al., 1995), BIOVIA Discovery Studio (Tabrez et al., 2021), SCFBIO (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp), adsorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction (http://lmmd.ecust.edu.cn/admetsar2), and AMBER20 (Case et al., 2020).
Preparation of proteins and ligands
Protein macromolecules were separated from the solvent and native ligands or nonstandard residues. Separation of the macromolecules from unnecessary molecules was carried out using the AutoDock Tools program. Then the optimization was conducted by adding polar hydrogen atoms and setting the grid box to the structures. The grid box was set up using the AutoLigand feature on AutoDock, to determine the position of the ligan-binding site according to the lowest energy obtained from the scanning (Harris and Goodsell, 2012). These results were saved in the PDBQT format. Finally, protein binding sites were visualized with the VMD software.
The ligands used were 20 natural compounds of kaurene diterpenoids. In addition, favipiravir and remdesivir that had been used for COVID-19 therapy were also used for comparison. The compound structures obtained from PubChem were converted into the PDB format using Open Babel and then optimized with AutoDock Tools. These results were saved in PDBQT format.
Molecular docking
AutoDock Vina was employed to attach kaurene diterpenoid compounds to the target proteins prepared and optimized with AutoDock Tools. At this stage, the value of the Gibbs free energy (ΔGL)/binding energy of each ligand to the target protein was obtained. Then the ligands were ranked based on the five lowest energy values. The ligands that produced the lowest energy in each protein were then observed for detailed interactions, including hydrogen bonds, hydrophobic interactions, alkyl, and the van der Waals interactions formed between the target protein and kaurene with LigPlot+ and BIOVIA Discovery Studio.
 | Table 1. List of kaurene diterpenoids and viral drugs as inhibitors of NSPs (3CLpro PLpro) and structural proteins (M, N) of SARS-CoV-2 used in this docking study. [Click here to view] |
MD simulations studies
AMBER20 conducted MD simulations on complexes ligand-protein with a total period of 50 ns simulation for each complex at a constant temperature of 300 K. MD simulation steps include minimization, heating, equilibration, and production run. MD simulations were carried out to verify docking results and analyze potential compounds’ binding behavior and stability in terms of root mean square deviation (RMSD) and root mean square fluctuation (RMSF). In addition, the coordinates of the structure were stored and examined using the CPPTRAJ package (Madej and Walker, 2020).
Pharmacokinetic properties
The drug-likeness analysis was carried out based on Lipinski’s rule of five (Lipinski, 2004) using SCFBIO (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp) (Jayaram et al., 2012). The ADMET study was done by using ADMET prediction (http://lmmd.ecust.edu.cn/admetsar2) (Rafi et al., 2020).
RESULTS AND DISCUSSION
Molecular docking is one of the most commonly utilized methods in structure-based drug design because of its capacity to predict the conformation of small-molecule ligands within the appropriate target binding site with a high degree of accuracy. Scoring functions are used in molecular docking systems to assess the binding energetics of the predicted ligand-receptor complexes. The energy variation due to the formation of the ligand-receptor complex is given by the binding constant (Kd) and the Gibbs free energy (ΔGL) (Ferreira et al., 2015).
The binding affinities of 20 natural compounds of kaurene diterpenoids to the nonstructural proteins (PLpro, 3CLpro, and helicase) and structural proteins (membrane, envelope, and nucleocapsid) of SARS-CoV-2 were estimated based on their estimated ligand binding energy/Gibbs free energy (ΔGL). The ligand with the lowest energy score had the highest protein-ligand binding affinity, while those with higher energy values had lower binding affinities (Subbaiyan et al., 2020). Therefore, the ligand with the highest affinity can be chosen as the potential drug for further studies.
This study also conducted a docking analysis of two medicines for COVID-19 therapy, favipiravir and remdesivir, for comparison. Remdesivir is a nucleotide analog inhibitor of the Ebola virus (EBOV) RdRp. Preliminary data showed a 33% mortality rate in 499 patients treated with remdesivir for the EBOV illness in the early stages of infection and during the same pandemic era, 75% fatality rate (almost 1,900 persons) of nontreated infected patients (Dyer, 2019). Remdesivir is effective against 2019-nCoV in Vero E6 cells (EC90 1.76 µM) (Wang et al., 2020). Favipiravir is a nucleoside analog, broad-spectrum antiviral drug (EC50 of 61.88 µM against SARS-CoV-2) and low toxicity (IC50 > 400 µM) (Viveiros Rosa and Santos, 2020).
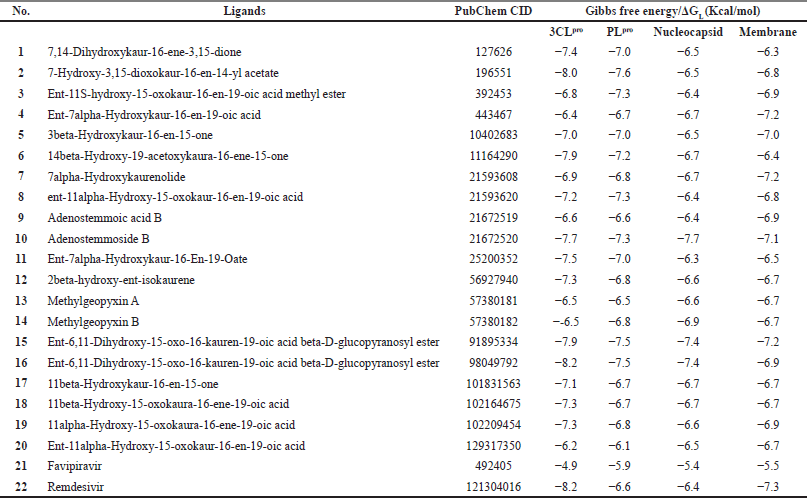
From the docking analysis, natural compounds of kaurene diterpenoids have a different protein-ligand binding affinity (Table S1). The five ligands with the lowest Gibbs free energy (ΔGL) for each receptor protein are presented in Table 2. The ligand that produced the lowest energy in each protein was then observed in the protein-ligand interaction (hydrogen bonds, hydrophobic interactions, alkyl, and the van der Waals interactions) with LigPlot+ and BIOVIA Discovery Studio.
Inhibition of 3-chymotrypsin-like cysteine protease (3CLpro) by kaurene diterpenoids
Upon transcription of the genome, beta-coronaviruses typically produce an 800 kDa polypeptide. Proteolytic cleavage of this polypeptide generates a variety of proteins. The proteolytic processing is mediated by 3-chymotrypsin-like cysteine protease (3CLpro) and papain-like protease (PLpro). The 3CLpro cleaves the polyprotein at 11 different sites, resulting in various NSPs essential for viral replication (Anand et al., 2014). Therefore, it is a potential target for anticoronaviruses inhibitors screening (Needle et al., 2015).
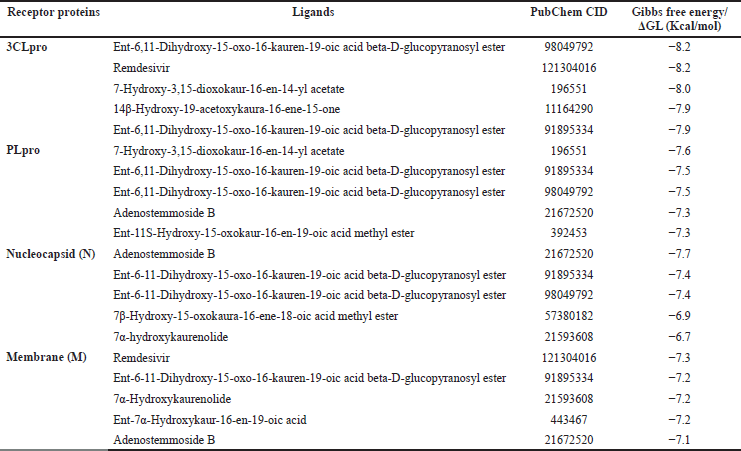 | Table 2. The binding energy of the top five kaurene diterpenoids with receptor proteins. [Click here to view] |
From Table 2, CID98049792 and remdesivir have an equal binding affinity with 3CLpro (−8.2 Kcal/mol), followed by other kaurene diterpenoid compounds (−8.0 until −6.2 Kcal/mol), and then favipiravir (−4.9 Kcal/mol). Before docking simulation, the target site for 3CLpro protein was chosen based on the location of cocrystallized inhibitor N3, which was thought to be the most commanding site for ligand interaction (Murugan et al., 2021). The binding pattern of CID98049792 with 3CLpro may impede substrate accessibility and subsequent inhibition, as demonstrated in Figure 1A. The strong hydrogen bond formed with Asn142 and His163 with bond lengths of 2.91 and 2.94 (Å), respectively. It has been observed that residues of 3CLpro are showing significant interaction, such as Arg188, Asp187, Tyr54, His164, Phe140, Ser144, Cys145, Leu141, and Gly143 (van der Waals interaction); His41, Met49, and Met165 (alkyl interaction); and Gln189, Glu166, and His172 (carbon-hydrogen bond interaction) as shown in Figure 1B and C. Those receptor-binding sites are also reported as 3CLpro SARS-CoV-2-natural compound binding sites (He et al., 2020). 3CLpro has catalytic residues Hys41 and Cys145 in the active site responsible for its enzymatic activity (Rahman et al., 2021). CID98049792 indicates a moderate binding was forming alkyl and van der Waals interaction with Hys41 and Cys145 residues located in between domains 1-2 of the 3CLpro.
Inhibition of papain-like protease (PLpro) by kaurene diterpenoids
Another important viral protease enzyme in SARS-CoV-2 replication is a papain-like protease (PLpro). Although PLpro is responsible for proteolysis, the host’s innate immunity antagonist, and viral replication (Rajpoot et al., 2021), it is an important target for antiviral medicines. Thus, the ligands against these enzymes can be suggested as potential drugs for COVID-19. The binding affinity of almost kaurene diterpenoid compounds with PLpro was relatively high (−7.6 until −6.7 Kcal/mol). However, surprisingly remdesivir and favipiravir have a lower binding affinity/high Gibbs free energy, −6.6 and −5.9, respectively (Table 2).
CID196551 has the lowest Gibbs free energy and showed good binding to the catalytic pocket of SARS-CoV-2 papain-like protease (PLpro) (Fig. 2A). CID196551 shows strong binding to PLpro through a hydrogen bond with Tyr273 (bond length 3.23 Å) and other significant and hydrophobic interactions via Asp302, Ser245, Thr301, Tyr264, Tyr268, Pro248, Pro247, and Met208 (Fig. 2B and C). The active site of PLpro consists of residues Glu167, Met208, Ala246, Asp164, Val165, Arg166, Pro247, Pro248, Tyr264, Cys217, Gly271, Tyr273, Thr301, Gly266, Asn267, Tyr268, Gln269, and Asp302. These active residues are present in the S3/S4 pockets of the Plpro palm domain (Arya et al., 2020). The active residue such as Tyr273 forms a hydrogen bond with CID196551, while Tyr264 and Pro248 form alkyl and pi-alkyl interactions, and other active residues Asp302, Thr301, Tyr268, Pro247, and Met208 form van der Waals interactions.
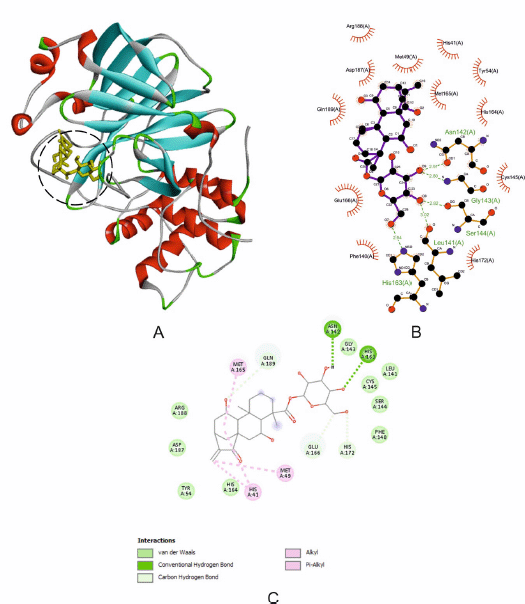 | Figure 1. The interaction of 3CLpro with CID98049792. (A) Blocking of protein catalytic residues. (B) The 3D binding mode of the complex. (C) The 2D binding mode of the complex. [Click here to view] |
Inhibition of nucleocapsid (N) protein by kaurene diterpenoids
A crucial phase in the virus’s life cycle is packaging its genome into new virions; with its large ~30 kb RNA genome, SARS-CoV-2 faces a difficult task. The SARS-CoV-2 nucleocapsid (N) protein (~45—60 kDa) is a structural protein responsible for binding, compacting, and packaging the viral genome (Lu et al., 2021; Ye et al., 2020). Beta-coronavirus N proteins also have a role in the viral RNA transcription of subgenomic mRNAs (Sawicki et al., 2007). The phase separation of the SARS-CoV-2 N protein with RNA, crucial for viral RNA packaging during virion development, reveals a new step in the viral life cycle that viral drugs could target. The binding affinity with the N protein of some kaurene diterpenoids is higher than remdesivir and favipiravir; in Table 2, we presented the top five compounds with the highest binding affinity (the lowest Gibbs free energy). Interaction between these compounds with the N protein may have an effect to disrupt the viral cycle.
CID21672520 exhibits good binding with the SARS-CoV-2 N protein (Fig. 3A) through two hydrogen bonds with N protein residues with a bond length (Å): Ile130 (2.98) and Ala134 (2.78; 3.05; 2.65) and other significant and hydrophobic interactions through residues Asn126, Ala125, Gly124, Lys127, Ile131, Tyr123, Gly69, Thr135, Gln70, Val133, Pro67, Phe66, and Trp132 as shown in Figure 3B and C. Hydrogen bonds formed between oxygen atoms in glycoside with oxygen and nitrogen atoms in amino acid residues. Significant hydrophobic interactions between CID196551 and N protein residues were carbon-hydrogen bond, van der Waals interaction, and alkyl interaction. Inhibition of adenostemmoside against protein N residues indicates that CID196551 can inhibit nucleocapsid self-assembly or mediate aberrant assembly of a new SARS-CoV-2 viral. Thus, it may be effective in reducing the severity of the infection and the patient’s infectivity.
Inhibition of membrane (M) protein by kaurene diterpenoids
The membrane (M) protein (~25—30 kDa) is essential for virus assembly, turning cellular membranes into workshops where virus and host factors combine to create new virus particles. Rigidity, spike clusters, and a relatively narrow range of membrane curvature are all related to the elongated M protein. On the contrary, the compact M protein is associated with ?exibility and low spike density (Neuman et al., 2011). The N-RNA and N-M interactions have an important contribution in the SARS-CoV-2 genome packaged into developing new virions (Lu et al., 2021). Thus, parallel to the N protein, the inhibition of the M protein could be targeted to obstruct the viral life cycle. The kaurene diterpenoids, which have the highest binding affinity with the M protein after remdesivir (−7.3 Kcal/mol), were CID91895334, CID21593608, and CID443467. The binding energy of those compounds with the M protein shown by the Gibbs free energy value was −7.2 Kcal/mol followed by other kaurene diterpenoid compounds (−7.1 until −6.3 Kcal/mol) and then favipiravir (−5.5 Kcal/mol), listed in Table 2.
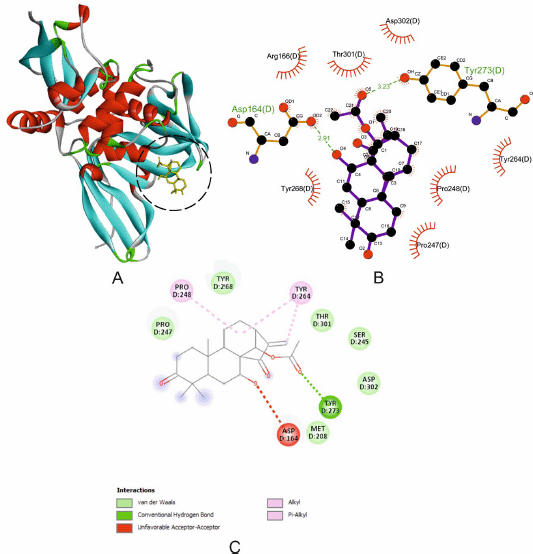 | Figure 2. The interaction of PLpro with CID196551. (A) Blocking of protein catalytic residues. (B) The 3D binding mode of the complex. (C) The 2D binding mode of the complex. [Click here to view] |
The three ligands that obtained the same Gibbs free energy (−7.2 Kcal/mol) were tested for their interaction with the SARS-CoV-2 M protein using BIOVIA and LigPlot+. However, only one ligand is shown here because it has the most hydrogen bonds and significant and hydrophobic interactions. The binding pattern of CID91895334 has suggested good binding to the pocket of the SARS-CoV-2 M protein, which may result in good inhibition of the SARS-CoV-2 M protein (Fig. 4A). The optimum binding of that ligand was formed through a hydrogen bond with residues Arg198, Glu137, Tyr196, and Thr77 with bond lengths (Å) 3.24, 2.94, 3.02, and 2.94, respectively. Other residues such as Gly78 and Val139 form carbon-hydrogen bonds, while Asn74, Leu138, Ile80, Ala81, Gly141, Phe65, Tyr179, Ser197, and Ala195 form van der Waals interactions, and residue Arg198 forms an alkyl interaction. The hydrogen bond and hydrophobic interactions are shown in Figure 4B and C. Viral RNA packaging such as SARS-CoV-2 is mediated first by the assembly of individual viral ribonucleoprotein complexes (vRNPs) along with the genomic RNA, followed by condensation of these RNPs and recruitment to developing virions through interactions with the M protein at the cytoplasmic side of the ER–Golgi intermediate compartment (Klein et al., 2020). Inhibition of the M protein residues by CID91895334 indicated this ligand could inhibit the process of SARS-CoV-2 RNA packaging. Therefore, the development of new SARS-CoV-2 virions can be disrupted.
The molecular docking study revealed that kaurene diterpenoids with the highest binding affinity to 3CLpro, nucleocapsid, and membrane of SARS-CoV-2 proteins were kaurene diterpene glycoside: CID98049792, CID91895334, and CID21672520. These three compounds have the same molecular structure but are different in chirality, as shown in Table 3. The highest binding affinity of these compounds may be due to a glycoside attached to the basic structure of the diterpene or aglycone. In glycoside, many oxygen atoms can form hydrogen bonds with oxygen and nitrogen atoms in amino acid residues on proteins and enzymes of SARS-CoV-2. This is evidenced by the ligand-protein interactions using BIOVIA and LigPlot+. Observation showed many hydrogen bonds in the active site residues with oxygen atoms in the glycosides of kaurene diterpenoids.
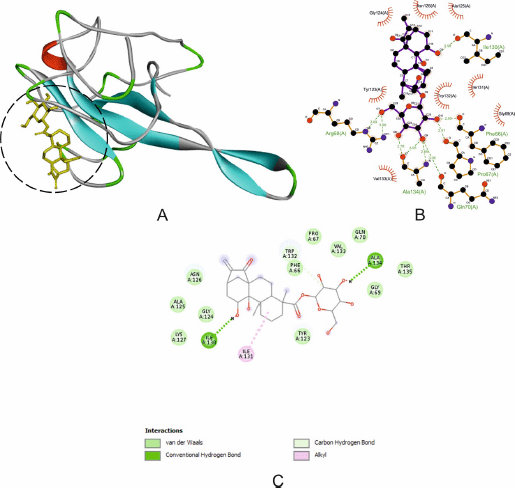 | Figure 3. The interaction of N protein with CID21672520. (A) Blocking of protein catalytic residues. (B) The 3D binding mode of the complex. (C) The 2D binding mode of the complex. [Click here to view] |
The inhibition of that kaurene diterpenoid on PLpro and 3CLpro is expected to inhibit the replication of the SARS-CoV-2 virus. Some natural products have been discovered to have antiviral activity by inhibiting viral replication ( Da Silveira Oliveira et al., 2017; Moghadamtousi et al., 2015). In this study, kaurene diterpene glycoside has inhibitory activity against replication enzymes and the structural proteins of SARS-CoV-2: nucleocapsid (N) and membrane (M). This finding suggested that kaurene diterpenoids also have inhibitory activity on viral assembly.
In general, a plant contains several kaurene diterpenoids. Adenostemma lavenia (L.) O. Kuntze with different local names, legetan warak (Indonesia), numa-daikon (Japan), xia tian ju or zhi ma hu (China and Taiwan), and vailenhlo (India), is traditionally used as a medicinal herb or traditional medicine to cure pneumonia, lung congestion, fever, hepatitis, and skin wounds (Cheng et al., 1979). Major phytochemicals reported from this plant are 11-hydroxylated kauranic acids (Cheng et al., 1979) and some glycosides, such as adenostemmosides A–G (Hamamoto et al., 2020; Shimizu et al., 1990). This docking study found that adenostemmoside B has a binding affinity to SARS-CoV-2 proteins nucleocapsid (N) and PLpro higher than antiviral drugs remdesivir and favipiravir. In this respect, a natural product such as A. lavenia contains several kaurene diterpenoids that can block SARS-CoV-2 virulence through inhibiting viral proteins such as 3CLpro, PLpro, N, and M, which are involved in the replication and assembly of SARS-CoV-2, unlike some synthetic antiviral drugs which can only prevent virus replication or inhibit further infection.
Molecular dynamic simulations
MD simulation was performed to evaluate the stability of the docked complexes using the AMBER software. The ligands with the lowest energy in each protein were observed at 50 ns in an explicit solvation system. A physiological salt concentration was also maintained. RMSD is the average displacement of atoms during simulation relative to a reference structure, usually the first frame of the simulation or crystallographic structure. RMSD is useful for the analysis of movement, depending on time and structure. It is often used to distinguish whether a structure is stable in the simulated timescale or deviates from the initial coordinates (Martínez, 2015).
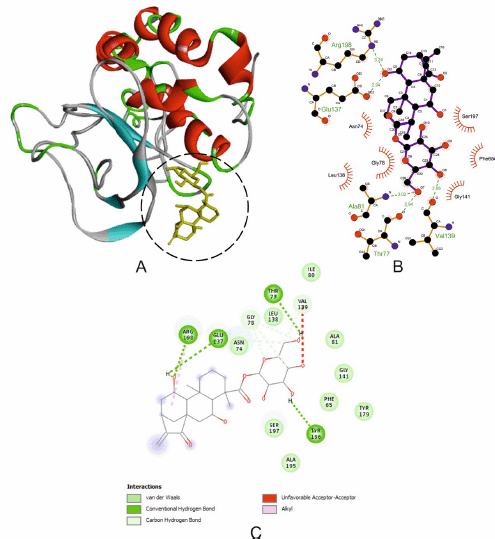 | Figure 4. The interaction of M protein with CID91895334. (A) Blocking of protein catalytic residues. (B) The 3D binding mode of the complex. (C) The 2D binding mode of the complex. [Click here to view] |
Based on the RMSD value (Table 4), the complexes between ligands and 3CLpro, Plpro, and nucleocapsid showed the small average values and a small minimum–maximum value range, indicating that each ligand is strongly bound to the binding site of proteins, resulting in minimal atomic movement from the initial position. The complex ligand-membrane has high average values and a high minimum–maximum value range, indicating their position changed the most. The stability of these protein-ligand complexes was analyzed using the RMSD of backbone atoms plotted against the total simulation time (Fig. 5A).
RMSF is the average deviation of each residue from the initial position during the simulation and indicates residual flexibility. Residue with a high RMSF value indicates that the residue is highly fluctuating or unstable to the initial conformation. On the other hand, if the RMSF value is low, the residue is stiffer to achieve stability (Kuzmanic and Zagrovic, 2010). Minimal fluctuations were observed for complexes between ligand and 3CLpro, and nucleocapsid remained stable throughout the simulations. At the same time, the complexes between ligand-PLpro and ligand-membrane showed a medium fluctuation of residues. However, these residues were not found at the active site. The RMSF of the ligands with the lowest energy in each target protein is shown in Figure 5B–E.
ADMET and pharmacokinetic properties
ADMET properties prediction and pharmacokinetics evaluation of pharmacological compounds provide critical information for determining and predicting whether a compound is suitable and reducing a potential risk in clinical development. Therefore, the ligands with the lowest Gibbs free energy (ΔGL) for each receptor protein were analyzed for ADMET and pharmacokinetics properties prediction. The ADMET and pharmacokinetics properties of these kaurene diterpenoids, remdesivir, and favipiravir are given in Table 5.
The ADMET prediction tool was used to predict the action of the drugs inside a host’s body. This study focused on human intestinal absorption, human oral bioavailability, carcinogenicity, Ames mutagenesis, and acute oral toxicity (LD50). The ADMET prediction tool exhibited that all kaurene diterpenoids, remdesivir, and favipiravir were well absorbed by the human intestine, noncarcinogenic compounds, and did not cause mutations in DNA. From the human oral bioavailability value, only favipiravir has a positive value indicating these compounds are well absorbed through oral administration. In contrast, all kaurene diterpenoids and remdesivir have a negative value, which means the optimum absorption is not through the oral route, maybe via the intravenous route. LD stands for lethal dose. LD50 is the amount of a substance given all at once that kills 50% (one-half) of a set of test animals. The LD50 is a method of determining a material’s acute toxicity (short-term poisoning potential). The acute oral toxicity (LD50) value showed that all kaurene diterpenoids (except CID196551), remdesivir, and favipiravir belonged to category III, which had an LD50 value of 500–5,000 mg/Kg. On the other hand, CID196551 has high acute oral toxicity because it was a category I (LD50 ≤ 50 mg/Kg).
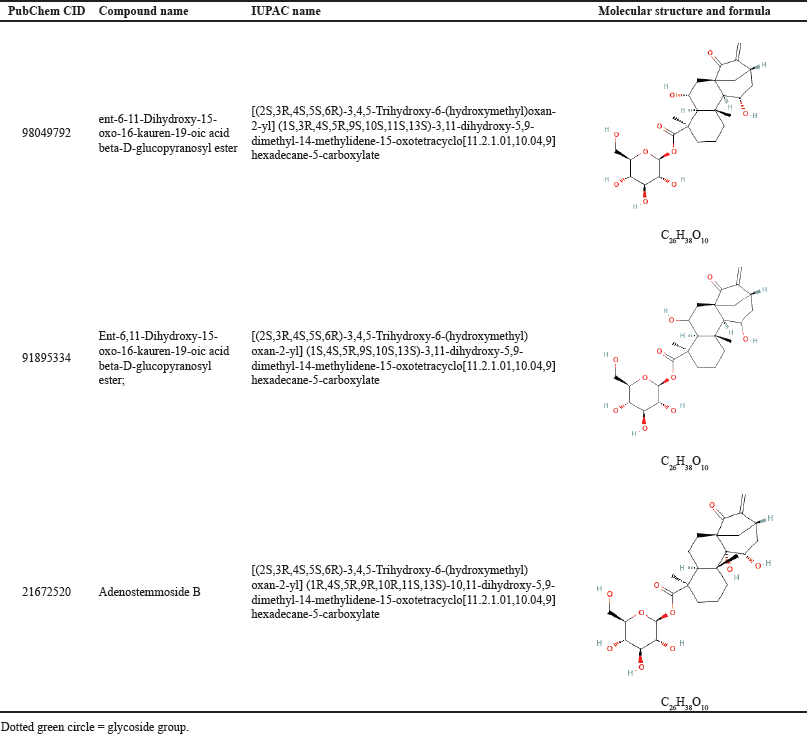 | Table 3. Kaurene diterpene glycosides have the highest binding affinity to 3CLpro, nucleocapsid, and membrane of SARS-CoV-2 proteins. [Click here to view] |
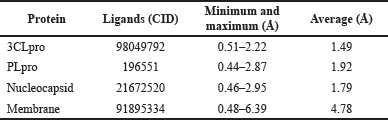 | Table 4. RMSD of the ligands with the lowest energy in each target protein. [Click here to view] |
Lipinski’s rule of five (Lipinski, 2004) is a rule to predict the pharmacological activity and probability of a drug being optimally absorbed orally in humans. The fact that CID196551 and favipiravir have a molecular mass <500 Daltons, H-bond donor < 5, and H-bond acceptors < 10 showed that these compounds could pass through the membrane and are well absorbed via the oral route. Meanwhile, CID98049792, CID91895334, CID21672520, and remdesivir are compounds with a large molecular mass > 500 Daltons, H-bond donor ≥ 5, and H-bond acceptors ≥ 10, and this indicates these compounds could not pass through the membrane and are well absorbed via the oral route. This result is in following the ADMET properties. All compounds have lipophilicity log P<5 and molar refractivity 40–130, except remdesivir > 130, which showed that all compounds could pass through the bilayer lipid on the cell membrane.
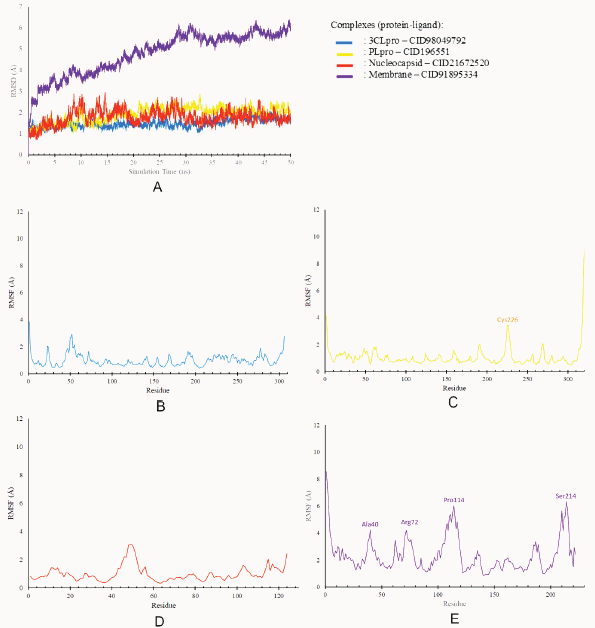 | Figure 5. Molecular dynamic analysis. (A) RMSD of protein-ligand complexes and RMSF of protein residues. (B) 3CLpro. (C) PLpro. (D) Nucleocapsid. (E) Membrane. [Click here to view] |
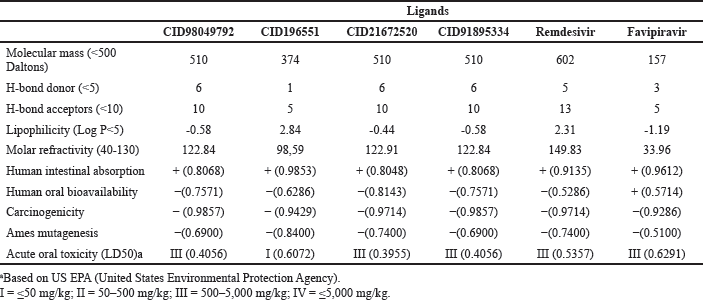 | Table 5. The ADMET and pharmacokinetics properties of kaurene diterpenoids, remdesivir, and favipiravir. [Click here to view] |
CONCLUSION
In this study, we performed computational studies to evaluate the activity of the kaurene diterpenoids as antivirals for the treatment of COVID-19. The molecular study exhibited the kaurene diterpene glycoside ent-6-11-dihydroxy-15-oxo-16-kauren-19-oic acid beta-D-glucopyranosyl ester (CID91895334 and CID98049792) and adenostemmosides B (CID21672520) have the inhibitory activity to 3CLpro, nucleocapsid, and membrane of SARS-CoV-2 proteins and have a higher binding affinity than other kaurene diterpenoids and even antiviral drugs such as remdesivir and favipiravir. This may be due to the presence of a glycoside that has many oxygen atoms that can form hydrogen bonds with oxygen and nitrogen atoms in amino acid residues on proteins and enzymes of SARS-CoV-2. Inhibition of these nonstructural and structural proteins has a significant impact on disrupting the viral replication and viral assembly of SARS-CoV-2. RMSD and RMSF analysis showed that almost all the complexes have minimum deviation and fluctuation, indicating that each ligand is strongly bound to the binding site of proteins and achieved stability. The ADMET prediction tool revealed that all kaurene diterpenoids were well absorbed by the human intestine, noncarcinogenic compounds, and did not cause mutations in DNA. We hope the findings of this study will be useful for developing novel natural anti-COVID-19 preventive and therapeutic agents. However, further in vivo and in vitro studies are obligatory to develop this potential inhibitor into a clinical medicine.
ACKNOWLEDGMENTS
This research and APC were funded by IPB University “Agromaritime Institution Research Assignment Grant FY 2021 No 7826/IT3.L1/PT.01.03/P/B/2021”. This research was supported by the bilateral exchange program directorate general of higher education/Japan society for the promotion of science joint research project 2021 No 103/E4.4/KU/2021.
AUTHORS’ CONTRIBUTIONS
All authors have made significant contributions to this manuscript. IB and STW contributed to the concept and design of the study. FA and NN contributed to data acquisition and analysis/interpretation. NN contributed to drafting the manuscript. IB, STW, and NN contributed to the critical revision of the manuscript. IB supervised the study and gave final approval. The published version of the manuscript has been read and approved by all authors.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Anand K, Anand K, Ziebuhr J, Wadhwani P. Coronavirus main protease (3CL pro) structure: basis for design of Anti-SARS drugs. Science, 2014; 300(5626):1763–7 CrossRef
Arashkia A, Jalilvand S, Mohajel N, Afchangi A, Azadmanesh K, Salehi-Vaziri M, Fazlalipour M, Pouriayevali MH, Jalali T, Mousavi NSD, Roohvand F, Shoja Z; SARS CoV-2 Rapid Response Team of Pasteur Institute of Iran (PII). Severe acute respiratory syndrome-coronavirus-2 spike (S) protein based vaccine candidates: state of the art and future prospects. Rev Med Virol, 2021; 31(3):1–15 CrossRef
Arya R, Das A, Prashar V, Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv, 2020 CrossRef
Batubara I, Astuti RI, Prastya ME, Ilmiawati A, Maeda M, Suzuki M, Hamamoto A, Takemori H. The antiaging effect of active fractions and ent-11α-hydroxy-15-oxo-kaur-16-en-19-oic acid isolated from Adenostemma lavenia (L.) O. Kuntze at the cellular level. Antioxidants, 2020; 9(8):719–33 CrossRef
Case DA, Walker RC, Cheatham TE, Wang CS, Roitberg A, Merz KM, Li P, Luo R, Darden T, Sagui C, Pan F, Wang J, Roe DR, LeGrand S, Swails J, Götz AW, Smith J, Cerutti D, Lee T, York D, Luchko T, Wilson L, Krasny R, Man V, Cruzeiro VWD, Monard G, Miao Y, Wang J, Cisneros GA, Duke RE, Skrynnikov NR, Mikhailovskii O, Xue Y, Izmailov SA, Kasavajhala K, Belfon K, Shen J, Harris R, Lin C, Onufriev A, Izadi S, Xiong Y, Wolf RM, Wu X, Gohlke H, Schott-Verdugo S, Qi R, Giambasu G, Liu J, Nguyen H, Brozell SR, Kovalenko A, Gilson M, Ben-Shalom I, Kurtzman T, Pantano S, Kollman PA.Amber 2020 Reference Manual. University of California, San Francisco, CA, 2020.
Chen JJ, Deng JS, Huang CC, Li PY, Liang YC, Chou CY, Huang GJ. P-coumaric-acid-containing Adenostemma lavenia ameliorates acute lung injury by activating AMPK/Nrf2/HO-1 signaling and improving the anti-oxidant response. Am J Chin Med, 2019; 47(7):1–24. CrossRef
Cheng PC, Hufford CD, Doorenbos NJ. Isolation of 11-hydroxylated kauranic acids from Adenostemma lavenia. J Nat Prod, 1979; 42(2):183–6 CrossRef
Coval SJ, Cross S, Bernardinelli G, Jefford CW. Brianthein V. A new cytotoxic and antiviral diterpene isolated from Briareum asbestinum. J Nat Prod, 1988; 51(5):981–4 CrossRef
Diniz LRL, Da Silva Maia Bezerra Filho C, Fielding BC, De Sousa DP, Gil G. Natural antioxidants: a review of studies on human and animal coronavirus. Oxid Med Cell Longev, 2020; 1–14 CrossRef
Dyer O. Two Ebola treatments halve deaths in trial in DRC outbreak. BMJ (Clin Res Ed), 2019; 366:1
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules, 2015; 20(7):13384–421. CrossRef
Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, Haagmans BL, Lauber C, Leontovich AM, Neuman BW,Penzar D, Perlman S, Poon LLM, Samborskiy D, Sidorov IA, Ziebuhr J. Severe acute respiratory syndrome-related coronavirus: the species and its viruses—a statement of the Coronavirus Study Group. bioRxiv, 2020; 1–15 CrossRef
Guo Y-R, Cao Q-D, Hong Z-S, Tan Y-Y, Chen S-D, Jin H-J, Tan K-S, Wang D-Y, Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Mil Med Res, 2020; 7(11):1–10 CrossRef
Guruprasad L. Human SARS CoV-2 spike protein mutations. Proteins Struct Funct Bioinform, 2021; 89(5):569–76 CrossRef
Hamamoto A, Isogai R, Maeda M, Hayazaki M, Horiyama E, Takashima S, Koketsu M, Takemori H. The high content of ent-11α-hydroxy-15-oxo-kaur-16-en-19-oic acid in Adenostemma lavenia (L.) O. Kuntze leaf extract: with preliminary in vivo assays. Foods, 2020; 9(73):1–12 CrossRef
Harris RM, Goodsell DS. Using AutoLigand with AutoDockTools. The Scripps Research Institute, La Jolla, CA, 2012.
He J, Hu L, Huang X, Wang C, Zhang Z, Wang Y. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs?: insights from structures of protease and inhibitors. Int J Antimicrob Agents, 2020; 56:1–10; doi:10.1016/j.ijantimicag.2020.106055 CrossRef
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph, 1996; 14(1):33–8 CrossRef
Islam MT, Sarkar C, El-Kersh DM, Jamaddar S, Uddin SJ, Shilpi JA, Mubarak MS. Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phytother Res, 2020; 34(10):2471–92 CrossRef
Jayaram B, Singh T, Mukherjee G, Mathur A, Shekhar S, Shekhar V. Sanjeevini: a freely accessible web-server for target directed lead molecule discovery. BMC Bioinform, 2012; 13(Suppl 17) CrossRef
Kim JH, Kismali G, Gupta SC. Natural products for the prevention and treatment of chronic inflammatory diseases: integrating traditional medicine into modern chronic diseases care. Evid Based Complement Altern Med, 2018 CrossRef
Kim KJ, Liu X, Komabayashi T, Jeong S Il, Selli S. Natural products for infectious diseases. Evid Based Complement Altern Med, 2016 CrossRef
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res, 2019; 47(D1):D1102– D1109 CrossRef
Klein S, Cortese M, Winter SL, Wachsmuth-Melm M, Neufeldt CJ, Cerikan B, Stanifer ML, Boulant S, Bartenschlager R, Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun, 2020; 11(1):1–10 CrossRef
Kuzmanic A, Zagrovic B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys J, 2010; 98(5):861–71 CrossRef
Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal, 2020; 10(2):102–8 CrossRef
Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol, 2004; 1(4):337–41; doi:10.1016/j.ddtec.2004.11.007 CrossRef
Lu S, Ye Q, Singh D, Cao Y, Diedrich JK, Yates JR, Villa E, Cleveland DW, Corbett KD. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun, 2021; 12(1):1–15 CrossRef
Lu Y, Wu K, Li L, He Y, Cui L, Liang N, Mu B. Characterization and evaluation of an oral microemulsion containing the antitumor diterpenoid compound ent-11alpha-hydroxy-15- oxo-kaur-16-en-19-oic-acid. Int J Nanomed, 2013; 8:1879–86 CrossRef
Madej BD, Walker R. AMBER Tutorial B0: an introduction to molecular dynamics simulations using AMBER, 2020.
Maier HJ, Bickerton E, Britton P. Coronaviruses: methods and protocols. 2015; 1282(1):1–282
Martínez L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS One, 2015; 10(3):1–10 CrossRef
Moghadamtousi SZ, Nikzad S, Kadir HA, Abubakar S, Zandi K. Potential antiviral agents from marine fungi: an overview. Mar Drugs, 2015; 13(7):4520–38 CrossRef
Morris GM, Goodsell DS, Pique ME, Lindstorm W, Huey R, Forli S, Hart WE, Halliday S, Belew R, Olson AJ. AutoDock version 4.2—user guide. Department of Molecular Biology, Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, 2014.
Murugan NA, Pandian CJ, Jeyakanthan J. Computational investigation on andrographis paniculata phytochemicals to evaluate their potency against SARS-CoV-2 in comparison to known antiviral compounds in drug trials. J Biomol Struct Dyn, 2021; 39(12):4415–26 CrossRef
Needle D, Lountos GT, Waugh DS. Structures of the middle east respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr D Biol Crystallogr, 2015; 71:1102–11 CrossRef
Neuman BW, Kiss G, Kunding AH, Bhella D, Baksh MF, Connelly S, Droese B, Klaus JP, Makino S, Sawicki SG, Siddell SG, Stamou DG, Wilson IA, Kuhn P, Buchmeier MJ. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol, 2011; 174(1):11–22 CrossRef
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open This research and APC were funded by IPB University “Agromaritime Institution Research Assignment. J Cheminform, 2011; 3(33):1–14 CrossRef
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, Masciovecchio C, Angeletti S, Ciccozzi M, Gallo RC, Zella D, Ippodrino R. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med, 2020; 18(1):1–9 CrossRef
Rafi MO, Bhattacharje G, Al-Khafaji K, Taskin-Tok T, Alfasane MA, Das AK, Parvez MAK, Rahman MS. Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: analogues of lopinavir and favipiravir as potential drug candidates against COVID-19. J Biomol Struct Dyn, 2020; 1–20 CrossRef
Rahman F, Tabrez S, Ali R, Alqahtani AS, Ahmed MZ, Rub A. Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. J Tradit Complement Med, 2021; 11(2):173–9 CrossRef
Rajpoot S, Alagumuthu M, Baig MS. Dual targeting of 3CLpro and PLpro of SARS-CoV-2: a novel structure-based design approach to treat COVID-19. Curr Res Struct Biol, 2021; 3:9–18 CrossRef
Saeed AAM, Ali AM, Fdhel TA. HPLC-ESI-MS analysis of some bioactive substances in two yemeni medicinal plants. Electron J Univ Aden Basic Appl Sci, 2020; 1(4):225–35; doi:10.47372/ejua-ba.2020.4.60 CrossRef
Sawicki SG, Sawicki DL, Siddell SG. A contemporary view of coronavirus transcription. J Virol, 2007; 81(1):20–9 CrossRef
Shimizu S, Miyase T, Umehara K, Ueno A. Kaurane-type diterpenes from Adenostemma lavenia O. Kuntze. Chem Pharm Bull, 1990; 38(5):1308–12 CrossRef
Da Silveira Oliveira AFC, Teixeira RR, De Oliveira AS, De Souza APM, Da Silva ML, De Paula SO, Schmidt TJ. Potential antivirals: natural products targeting replication enzymes of dengue and chikungunya viruses. Molecules, 2017; 22:505 CrossRef
Subbaiyan A, Ravichandran K, Singh SV, Sankar M, Thomas P, Dhama K, Malik YS, Singh RK, Chaudhuri P. In silico molecular docking analysis targeting SARS-CoV-2 spike protein and selected herbal constituents. J Pure Appl Microbiol, 2020; 14(1):989–98 CrossRef
Tabrez S, Rahman F, Ali R, Alouffi AS, Akand SK, Alshehri BM, Alshammari FA, Alam A, Alaidarous MA, Banawas S, Dukhyil AAB, Rub A . Cynaroside inhibits leishmania donovani UDP-galactopyranose mutase and induces reactive oxygen species to exert antileishmanial response. Biosci Rep, 2021; 41(1):1–14 CrossRef
Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010; 31(2):455–61; doi:10.1002/jcc.21334 CrossRef
Viveiros Rosa SG, Santos WC. Clinical trials on drug repositioning for COVID-19 treatment. Rev Panam Salud Publica/Pan Am J Public Health, 2020; 44:1–7 CrossRef
Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel, 1995; 8(2):127–34 CrossRef
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res, 2020; 30(3):269–71 CrossRef
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B, 2020a; 10(5):766–88 CrossRef
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020b; 579(7798):265–269 CrossRef
Xiantian X, Ping C, Jingfang W, Jiannan F, Hui Z, Xuan L, Wu Z, Pei H. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci, 2020; 63(3):457–60 CrossRef
Ye Q, West AMV, Silletti S, Corbett KD. Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Protein Sci, 2020; 29(9):1890–901 CrossRef
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science, 2020; 368(6489):409–12 CrossRef
SUPPLEMENTARY MATERIAL
 | Table S1. The binding energy of ligands (kaurene diterpenoids) with receptor proteins. [Click here to view] |