
INTRODUCTION
Hypercalciuria is the most usual metabolic disease that occurs in approximately 50% of nephrolithiasis patients, while hyperoxaluria is found in 10%–50% of urinary calculus cases (DiBianco et al., 2015). Hypercalciuria is a condition in which the urine calcium concentration is >8 mmol/day (European Association of Urology, 2018). Hyperoxaluria is a condition where the urine oxalate concentration exceeds 40 mg/day or 0.5 mmol/day (European Association of Urology, 2018; Figueres et al., 2020). Hypercalciuria and hyperoxaluria can be diagnosed by determining the calcium and oxalate levels in the patient’s urine and blood. Both diseases are included in the metabolic syndromes caused by various pathological conditions (Arrabal-Polo et al., 2012; Witting et al., 2021).
The pathophysiology of hypercalciuria is related to a defect in the reabsorption of renal tubular calcium, an increase in the absorption of intestinal calcium, and the reabsorption of minerals in bone (García Nieto et al., 2019). Hyperoxaluria can be generated by inherited disorders, intestinal disease, or dietary oxalate consumption (Witting et al., 2021). A disparity of calcium and oxalate levels in the blood can lead to the precipitation of calcium oxalate which is crystalized as the nephroliths. Subtypes of calcium oxalate are more commonly found than calcium phosphate (4:1 in ratio) (Ivanovski and Drüeke, 2013).
The goal of therapy for hypercalciuria and hyperoxaluria is to reduce the excretion of calcium and oxalate (European Association of Urology, 2018). There are some conditions where the treatment cannot reduce the excretion of calcium and oxalate and causes comorbidities such as kidney stones. These conditions result in the implementation of pharmacotherapy for hypercalciuria and hyperoxaluria and this needs to be studied for its application in cases based on reducing excess calcium and oxalate excretion and also prevention and improvement of stone conditions in patients. Therefore, a study to support the best therapy for hypercalciuria and hyperoxaluria is required due to their various conditions and associated diseases. This paper aims to review hypercalciuria and hyperoxaluria by their pathophysiology, their association with the mechanism of urinary calculus development, and the pharmacotherapy by examining several case reports.
METHODS
The articles used in this study were traced through two databases, PubMed and Google Scholar, within 20 years of the publication date. The literature search on the PubMed database was carried out from June 2021 to November 2021 using advanced searches and the Boolean operators “AND,” “OR,” and “NOT”: (((((Pathophysiology) OR (Calcium Homeostasis) OR (Diagnosis)) OR (Pharmacotherapy)) OR (Treatment) AND (Hypercalciuria)) OR (Hyperoxaluria)) NOT (Hypercalcemia) NOT (hypocalcemia). A literature search on the Google Scholar database was carried out using the following advanced search: Calcium Oxalate Stone Formation OR Pathophysiology OR Diagnosis OR Pharmacotherapy OR Treatment “Hypercalciuria Hyperoxaluria” —Hypercalcemia-hypocalcemia-in vivo-in vitro. The articles obtained from the initial search were 640 studies. Articles published before 2001, non-English studies, nonclinical researches, and unrelated studies that do not contain the content information were excluded; thus, only 54 articles were included in this study. This method has been summarized in Figure 1.
RESULTS AND DISCUSSION
Calcium regulatory mechanism
Calcium is responsible for various physiological functions, such as the metabolism of bone, conduction of nerves, contraction of muscle, and intracellular signaling (Felsenfeld et al., 2013). The total amount of calcium is 1,000–1,200 g in the human body. Approximately, 99% of the calcium is deposited in the bones while the rest is distributed in the extracellular and intracellular areas, which plays an important role as the extracellular and intracellular cation (Blaine et al., 2015). Specific extracellular calcium concentrations are maintained by absorption in the bowel, reabsorption in the kidneys, and resorption/formation in the bone (Felsenfeld et al., 2013; Pu et al., 2016). Calcium homeostasis has been summarized in Figure 2.
Intestinal calcium absorption
There are two ways in which calcium is absorbed from food by the intestine. These are transcellular and paracellular absorptions. Transcellular absorption activity of calcium (80%) occurs in the duodenum (Pu et al., 2016). The intracellular calcium-binding protein and calcium pump play an important role in transcellular calcium absorption. Transcellular calcium absorption is commenced by transient receptor potential vanilloid type 6 (TRPV6), a selective calcium channel receptor situated on the surface of the membrane, which opens for the flowing of calcium ions into the cells (Richards, 2006). Once calcium ions infiltrate the cell via TRPV6, it binds to a calcium buffer protein and is released from the cells into the circulation through the plasma membrane ATPase 1b (PMCA1b) and NCX1 (Christakos, 2012; Pu et al., 2016).
Calcium is mainly absorbed via paracellular absorption when the calcium level in the lumen elevates. This route is indirectly affected by calcitriol [1,25(OH)2D] in controlling calcium absorption. Calcium flows down its concentration gradient to the apical microvilli through calcium channels. The luminal calcium concentration is higher than intracellular calcium concentration; large gradient concentration supports the passive movement of calcium. Calcium is reversibly and quickly bound to the calmodulin-actin-myosin I complex. Calcium moves to the basolateral cell area via microvesicular transport. The ace-calmodulin actin-myosin I complex becomes saturated with calcium, and concentration gradient decreases and slows calcium absorption (Keller and Schinke, 2013).
Calcitriol works on the epithelial cells of the intestine to elevate the synthesis of calbindin (calcium-binding protein). Calcium is bound to calbindin and releases the calcium-calmodulin complex and then removes calcium from the microvilli area. The decreased concentration of calcium supports the calcium movement into the microvilli. When the complex between calcium and calbindin cleaves, calcium in the cell is actively free and ready to be exchanged with sodium (Blaine et al., 2015; Keller and Schinke, 2013).
Renal calcium reabsorption
The nephron proximal tubules of the kidneys function as the sensor for the reabsorption of calcium (Blaine et al., 2015). There are two pathways of calcium reabsorption in the kidneys which are the paracellular and transepithelial pathways. Calcium reabsorption in the transepithelial pathway in the kidneys consists of three main stages. In the first stage, calcium is delivered to the intracellular space via TRPV5. Eventually, calcium attaches to calcium-binding proteins, such as calbindin D28k and calbindin D9k, and then it passively propagates across the basolateral membrane due to its gradient. Finally, calcium is transported by PMCA1b and/or NCX1 to the blood (Pu et al., 2016).
Basal calcium absorption occurs through a paracellular pathway whose movement rate is regulated by the electrochemical driving force determined by the rate of sodium absorption. Approximately two-thirds of bulk reabsorption of sodium, chloride, water, and calcium occurs in the nephron proximal tubule. Concomitantly, the level of intraluminal chloride elevates higher than in the plasma. In addition, at the beginning of sodium transport, a negative lumen potential occurs. Gradient electrochemistry promotes paracellular chloride reabsorption in the proximal tubules which leads to a positive lumen potential (Curry and Yu, 2019).
Bone calcium resorption
Bone resorption plays a key role in modulating blood calcium levels. Bones are constantly being formed by osteoblasts and osteoclasts. Osteoblasts support bone formation, whereas osteoclasts facilitate bone tissue destruction and calcium release. Development and activation of osteoclast are mediated by the NF-B (RANK) and ligand-receptor activator (RANKL). The expression of RANKL is promoted by vitamin D3, parathyroid hormone (PTH), PTHrP, TNF-α, and interleukins and inhibited by TGF-β. Meanwhile, the activities of RANKL are also inhibited by osteoprotegerin (OPG). OPG interferes with RANK and RANKL interaction by competitively binding with RANKL and serves as a barrier to osteoclasts development and activation. The OPG level of expression in the bone marrow is inhibited by 1,25-dihydroxyvitamin D3 and PTH (Pu et al., 2016).
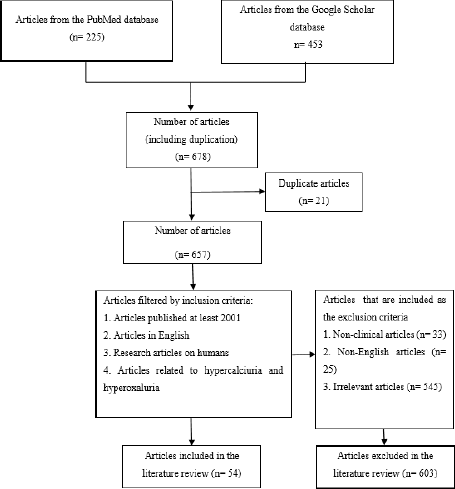 | Figure 1. Flowchart of literature searching. [Click here to view] |
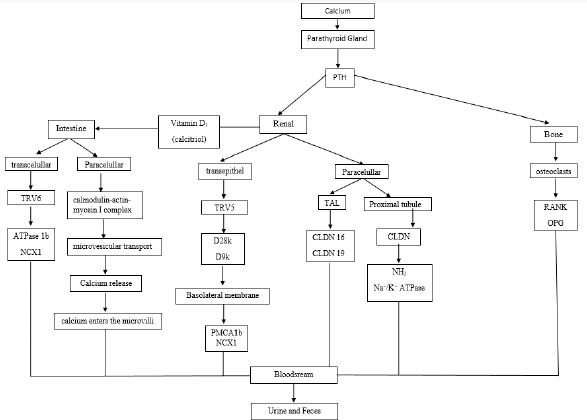 | Figure 2. Diagram of calcium homeostasis. [Click here to view] |
Oxalate regulatory mechanism
Oxalate originates from both endogenous and exogenous pathways. Endogenous oxalate is the metabolism end product, mostly found in the liver, and represents 85%–90% of the total oxalate in the blood and is excreted through bile secretions. Exogenous oxalate which represents about 10%–15% of the oxalate in the blood comes from food absorption in the digestive tract (Brzica et al., 2013). A small fraction (2%–10%) of the total amount of oxalate in the diet is absorbed. On the other hand, most is retained in the intestine. Daily oxalate intake in humans is about 44–352 mg/day, with an average intake of about 130–152 mg/day (Huang et al., 2018).
Endogenous oxalate is the hepatic metabolism end product that comes from two main sources, e.g., ascorbic acid and glyoxylic acid. Although the ascorbate acid source accounts for about 30% of total metabolism and excretion, the exact metabolic pathway needs to be further studied. The main source of oxalate is derived from the oxidizing reaction of glyoxylate. This metabolic reaction is catalyzed by several enzymes, including alcohol dehydrogenase 1, lactate dehydrogenase (LDH), acid hydroxy oxidase 1, xanthine oxidase, alanine glyoxylate aminotransferase (AGT), and glyoxylate reductase (GRHPR) (Burns et al., 2020).
The concentration of glyoxylate is mainly regulated by multiple pathways. Glyoxylate can metabolize by glycolate oxidase or LDH to become oxalate. Glyoxylates are important intermediate products in various reactions. Excess glyoxylate elevates its transformation to oxalate, which causes hyperoxaluria. AGT is facilitated by the conversion of glyoxylate and alanine to glycine and pyruvate through transamination. The full activity of AGT depends on the pyridoxine cofactor (vitamin B6). Glycine is converted to serine and serine to OH-pyruvate, which is metabolized by GRHPR. Subsequently, glycolaldehyde is metabolized to glyoxylate through several steps. The path source of another glyoxylate is via the cleavage of hydroxyproline via 4-hydroxy-2 oxoglutarate aldolase (HOGA1) (Brzica et al., 2013; Burns et al., 2020).
Exogenous oxalates are absorbed from food in the gastrointestinal tract (2%–10%), while most (90%–98%) are used as energy sources by intestinal bacteria or eliminated in the feces. Endogenous and exogenous oxalate contribute to total oxalate in the blood. The larger portion of the oxalate in the blood is excreted from the organism, mostly (90%–95%) in the kidney and the remainder in the intestines. A small portion goes back to the hepatocytes and continues to recycle with the enterohepatic circulation (Ermer et al., 2017).
Pathophysiology of hypercalciuria
Pathophysiology of hypercalciuria may associate with an increase of calcium transport in the intestine, renal tubule reabsorption, and the regulation of bone mineral content. The pathophysiology has been summarized in Figure 3.
Absorptive hypercalciuria
Absorptive hypercalciuria results from the activation of calcitriol in the gut which stimulates secondary absorption of active calcium via TRPV6. Increased gastrointestinal absorption of calcium leads to higher serum calcium as well as a decrease in serum vitamin D and PTH. Only a small quantity of ingested calcium is absorbed in the small intestine. Patients with absorptive hypercalciuria usually have relatively high serum vitamin D levels (Frick and Bushinsky, 2003; Pak et al., 2015). The cause of this abnormality can be idiopathic, hypervitaminosis D (iatrogenic, sarcoidosis, or genetic), or homozygous mutations in the CYP24A1 or SLC34A1/SLC34A3 genes. These genes encode for vitamin D 24-hydroxylase and the sodium/phosphate cotransporter. However, heterozygous mutations are difficult to detect because their interpretation is difficult in clinical practice (Figueres et al., 2020).
Renal hypercalciuria
Renal calcium loss might occur by two mechanisms in patients with hypercalciuria, by increasing filtered calcium in the glomerulus and by reducing reabsorbed calcium in the tubule. Calcium filtered loads do not increase with meals in normal individuals. On the other hand, the urinary calcium levels in idiopathic hypercalciuria exceed in the fasting state and are significantly higher than normal in the after-meal condition. The proximal tubule of the nephron plays an important role in increasing urine calcium excretion in hypercalciuria patients (Worcester et al., 2021).
Resorptive hypercalciuria
Resorptive hypercalciuria correlates with absorptive hypercalciuria. In resorptive hypercalciuria, there is an increase in bone resorption. Bone demineralization is an effect that occurs after several years. Resorptive hypercalciuria can occur due to pathological bone abnormalities that increase bone catabolism (Figueres et al., 2020; Zerwekh, 2008). Increased calcium uptake can increase urinary sodium excretion, increase tubular sodium load, and decrease tubular calcium reabsorption, thereby supporting a decrease in bone mineral density (BMD) over time (Ryan and Ing, 2021). Decreased BMD and increased incidence of fractures are increased in patients (Worcester et al., 2021). Resorptive hypercalciuria occurs in 3%–5% of cases of hypercalciuria. The most common cause of resorptive hypercalciuria is hyperparathyroidism. The abnormality of serum PTH that is secreted excessively causes the release of calcium from bone; this is an indication of osteopenia and hypercalcemia (Pak et al., 2015).
Idiopathic hypercalciuria
The pathophysiology of idiopathic hypercalciuria is complex and is not completely understood; this is because idiopathic hypercalciuria is a systemic disorder of calcium balance in the body with changes in ion transport in the intestine, kidney, and bone (Penido and Tavares, 2012). Idiopathic hypercalciuria can be defined as persistent hypercalciuria despite normal or limited calcium intake, or hypercalciuria with normal levels of PTH, phosphorus, 1,25-dihydroxy vitamin D (calcitriol), and serum calcium (Ryan and Ing, 2012). High sodium intake can increase urinary sodium excretion, and an increase in tubular sodium load causes a decrease in tubular calcium reabsorption, thereby reducing BMD (Vezzoli et al., 2014). Primary high sodium is rare in cases of total idiopathic hypercalciuria. High protein intake leads to an increase in dietary acid load and bone calcium and also inhibits renal calcium reabsorption. Increasing dietary protein from 0.5 to 2.0 mg/kg/day can increase urinary calcium output (Siener et al., 2005).
Pathophysiology of hyperoxaluria
The pathophysiology of hyperoxaluria includes AGT deficiency, GRHPR deficiency, HOGA defect, or increased intake of oxalic precursors. The pathophysiology of hyperoxaluria has been summarized in Figure 4.
Primary hyperoxaluria
Primary hyperoxaluria type 1 results from a deficiency of the liver-specific peroxisomal enzyme AGT. The action of the AGT enzyme depends on pyridoxal 5′-phosphate which catalyzes the transamination of glyoxylate to glycine. AGT deficiency causes the accumulation of glyoxylate and excessive production of oxalate and glycolate. AGT has an N-terminal amino acid wrapped around an adjacent monomer and is a stable homodimer. Pro11Leu is a common variant creating a more robust N-terminal mitochondrial targeting sequence, which influences the fate of several mutant proteins (Cochat and Rumsby, 2013).
Primary hyperoxaluria type 2 results from a deficiency of GRHPR, a catalyst that plays a role in the reduction of glyoxylate to glycolate and hydroxypyruvate to D-glycerate. GRHPR deficiency causes LDH to metabolize accumulated glyoxylate to oxalate and hydroxypyruvate to l-glycerate (Cochat and Rumsby, 2013).
Primary hyperoxaluria type 3 is caused by a defect in hepatic mitochondrial enzyme HOGA which functions in the metabolism of hydroxyproline with the forward reaction of 4-hydroxy-2-oxoglutarate (HOG) being converted into pyruvate and glyoxylate (Cochat and Rumsby, 2013). The increase in oxalate levels due to impaired glyoxylate synthesis is thought to be caused by HOG breaking down into oxalate enzymatically or in other ways or HOG inhibiting mitochondrial GRHPR (Belostotsky et al., 2012; Riedel et al., 2012).
Secondary hyperoxaluria
Secondary hyperoxaluria results from increased intake of oxalic precursors (ascorbic acid or ethylene glycol) or increased absorption of oxalate from food (Glew et al., 2014; Martines et al., 2019). Malabsorption causes calcium not to precipitate with oxalate but to bind with increased intraluminal free fatty acid levels. Unbound oxalate is then freely absorbed into the blood via passive paracellular colonic transport (Nazzal et al., 2016). The higher absorption of oxalate in the gastrointestinal tract and the production of oxalate in the liver lead to an elevation of plasma and urinary oxalate, which eventually increase the risk of nephrolithiasis (Curhan and Taylor, 2008). Small intestine malabsorption or excess of free fatty acids from the food results in increased fat delivery to the colon. Fats can bind with dietary calcium and increase absorbed free oxalate in the bloodstream (Witting et al., 2021).
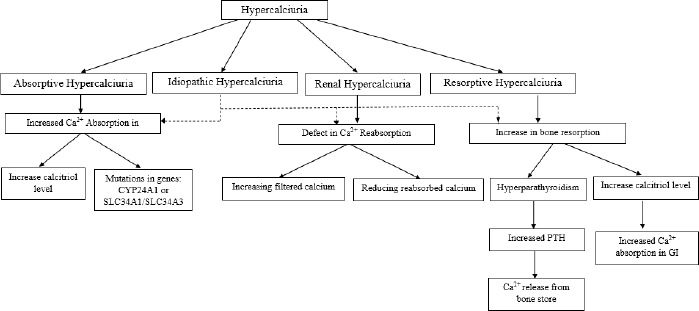 | Figure 3. Diagram of hypercalciuria pathophysiology. [Click here to view] |
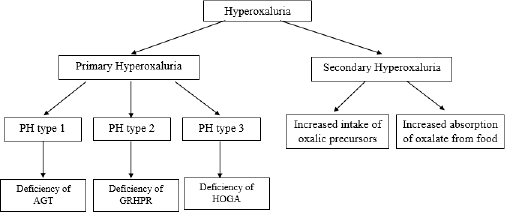 | Figure 4. Diagram of hyperoxaluria pathophysiology. [Click here to view] |
Mechanism of calcium oxalate stone formation
The formation of calcium oxalate stones is classified into calcium oxalate monohydrate, dihydrate, and trihydrate (Adnan and Nigwekar, 2019). Randall’s plaque is calcium plaque deposited in the interstitial tissue of the renal papillae that acts as a nidus for the formation of urinary tract stones. Plaque formation begins in the descending limb of the loop of Henle and extends to the medulla interstitium. The integrity of the urothelium will be damaged so that the plaque area is exposed to urine. Urine molecules, osteopontin, the Tamm-Horsfall protein, and crystals in supersaturated urine react with exposed plaques to produce alternating matrix bands and crystal layers with repeated layering and crystallization. Crystals extend from the modulated matrix outward into the urinary space and begin to form calcium oxalate stones (Chung, 2014). The amount of oxalate excreted in the urine is responsible for the formation of calcium oxalate stones (Mitchell et al., 2021).
Pharmacotherapy of hypercalciuria
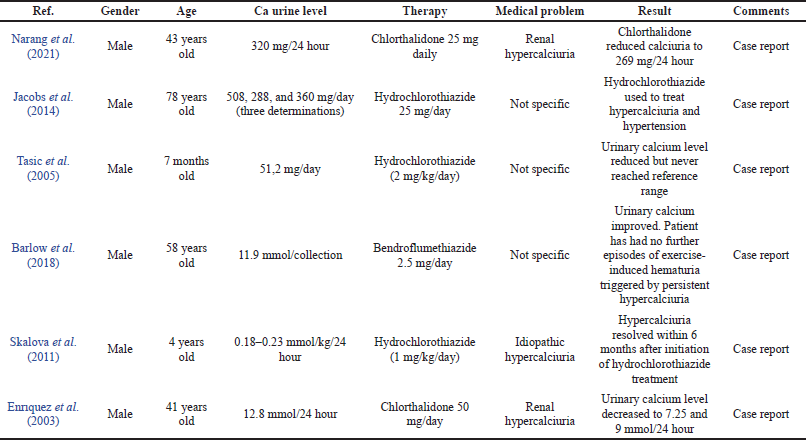
The six case studies in Table 1 show a description of the cases of hypercalciuria that occurred in male patients of varying ages. Reported types of hypercalciuria vary, namely, idiopathic hypercalciuria and renal hypercalciuria, and some are not specifically mentioned. Hydrochlorothiazide was used in two cases, chlorthalidone in two cases, and bendroflumethiazide in one case as shown in Table 1.
Hydrochlorothiazide
Hydrochlorothiazide is used in the treatment of hypercalciuria patients with variations in age and type of hypercalciuria. The results of therapy with hydrochlorothiazide in two cases in the elderly showed a decrease in 24 hours urinary excretions. This decrease reached its normal value within 6 months in a case report of a 4-year-old with idiopathic hypercalciuria (Skalova et al., 2011). However, in a case report of a 7-month-old with hypercalciuria, the decrease in 24 hours urinary calcium did not reach the normal value. This condition occurs because of the occurrence of CLDN16 mutations. CLDN16 may also play a role in pure calcium disturbances. Therefore, in these conditions, treatment should aim at reducing the complications of the disease because of the decrease in 24 hours urinary excretions (Tasic et al., 2005). The case of hypercalciuria in a patient aged 73 years using a dose of 25 mg/day indicated the treatment dose for hypercalciuria and a dose of 100 mg/day for the most severe hypertension (Jacobs et al., 2014).
Compared with the UEA Guidelines for Urolithiasis in Table 2, hydrochlorothiazide for hypercalciuria was used at 25–50 mg/day in adults and 0.5–1 mg/kg/day in children (European Association of Urology, 2018). Thiazides can increase calcium reabsorption in the proximal and distal tubule, resulting in a reduction of urinary calcium (Pak, 2008). The use of hydrochlorothiazide in pediatric patients aged 4 years at a dose of 1 mg/kg/day and in adults at 25 mg/day was in accordance with the dosage prescribed by the UAE Guidelines for Urolithiasis in hypercalciuria patients that showed a decrease in 24 hours urinary calcium up to 50% (European Association of Urology, 2018; Jacobs et al., 2014; Skalova et al., 2011). Moreover, a case reported a 7-month-old treated with doses above the standard UAE Guidelines for Urolithiasis for nonspecific reasons (Tasic et al., 2005).
 | Table 1. Use of pharmacotherapy in case reports of hypercalciuria. [Click here to view] |
Bendroflumethiazide
Bendroflumethiazide therapy was used in the case of a 58-year-old man with hypercalciuria with a history of carbonate apatite and urolithiasis. The patient experienced hematuria after high-intensity running due to the formation of lesions in the urothelial trauma secondary induced by persistent hypercalciuria. After the use of fluid intake and bendroflumethiazide 2.5 mg/day, there was an improvement in urinary calcium level and there was no continued hematuria (Barlow et al., 2018). The UEA Guidelines for Urolithiasis did not describe the use of bendroflumethiazide in the treatment of hypercalciuria, but bendroflumethiazide also reduced stone recurrence and is a drug with the same class and mechanism as hydrochlorothiazide (Dhayat et al., 2018).
Chlorthalidone
There are two cases attached to Table 1 on the treatment of hypercalciuria cases with chlorthalidone. The first case was a 53-year-old male patient with a new kidney since the age of 10 years, stage III CKD at the age of 17 years, and at the age of 43 metabolic abnormalities including hypercalciuria occurring. Treatment of hypercalciuria at 12.5 mg/day with hydrochlorothiazide was performed, but there was no significant improvement followed by the formation of kidney stones. Chlorthalidone was then given instead of hydrochlorothiazide, and ureteroscopic stone procedures were performed. There was a decrease in urinary calcium level from 320 to 269 mg/day; this decrease was closest to the normal value in 2 years of treatment (Narang et al., 2021). The second case was a 41-year-old man with a diagnosis of renal hypercalciuria with a history of kidney stone surgery at the age of 20. In this case, the patient had hypercalciuria and nephrolithiasis; the chlorthalidone regimen reduced urinary calcium levels and relieved symptoms (Enr?quez et al., 2003).
Chlortalidone at a dose of 25 mg/day is also recommended as an alternative treatment of the UAE Guidelines for Urolithiasis in hypercalciuria in adult patients. Chlortalidone has a longer half-life than hydrochlorothiazide so it can be given once a day (Pak, 2008). The decrease in 24 hours urinary calcium in the two cases reported in Table 1 was 16%–43%. Cases in renal hypercalciuria patients aged 43 used the same dose as the UAE Guidelines for Urolithiasis, which was 25 mg/day (Narang et al., 2021), but other cases reported the use of a higher dose than the UAE Guidelines for Urolithiasis (50 mg/day). However, this dose is accepted as a preventive therapy for kidney stones (Fontenelle and Sarti, 2019).
Pharmacotherapy of hyperoxaluria
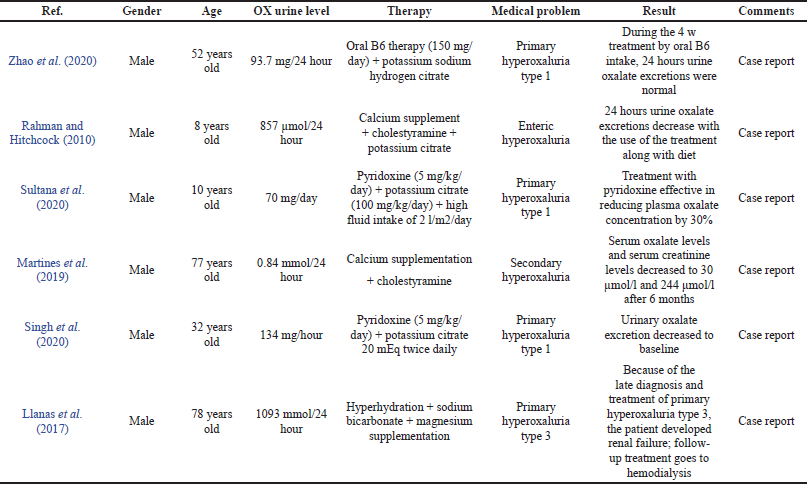
There are six cases presented in Table 3 with different cases and conditions of hyperoxaluria. The use of drugs, in general, is based on the classification of hyperoxaluria, namely, primary and secondary hyperoxaluria. The various therapeutic regimens attached to Table 3 include pyridoxine, alkaline citrate, magnesium supplementation, calcium supplementation, and other adjunctive therapies.
Pyridoxine
There are three studies that used pyridoxine in combination with other therapies to treat hyperoxaluria in male patients, aged 53 years and 10 years, with a diagnosis of primary hyperoxaluria type 1. A diagnosis of primary hyperoxaluria type 1 with mutations in the AGXT gene was confirmed in the first patient aged 53 years after allogeneic kidney transplantation. The use of pyridoxine showed sensitive results, and 24 hours urinary excretion begins after transplantation and then returns to normal values within 4 months (Zhao et al., 2020).
The second case was of a patient aged 10 years also with a previous history of bladder stones. This patient received treatment with peritoneal dialysis, but there was an increase in urinary oxalate levels concurrently before peritoneal dialysis so that hyperoxaluria treatment was carried out at the same time as the procedure. Laboratory results showed improvement in various parameters including serum oxalate, but the results of 24 hours urinary oxalate excretion were not specified (Sultana et al., 2020). The third case in a 32-year-old patient also showed sensitivity to pyridoxine with a decrease in urinary oxalate excretion from 297.5 mg/day to baseline (Singh et al., 2020).
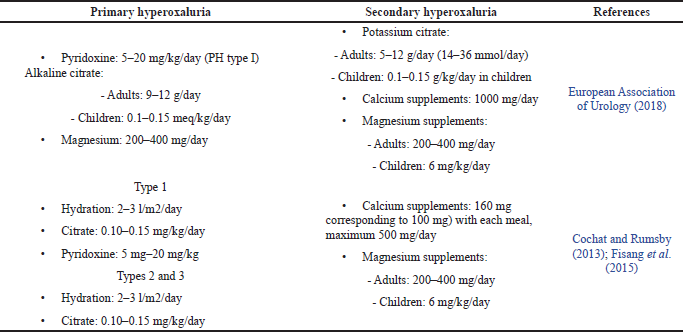
Compared with the UAE Guidelines for Urolithiasis, pyridoxine is given only to patients with primary type 1 hyperoxaluria. The dose of pyridoxine given depends on body weight in the range of 5–20 mg/kg/day. The use of pyridoxine aims to reduce urinary oxalate excretion. There are three cases of pyridoxine use in Table 3 with primary type 1 hyperoxaluria (European Association of Urology, 2018). Two cases used doses in the specified range, 5 mg/kg/day, with a decrease in urinary oxalate of 30%–70% with different treatment periods, but one case did not use units based on body weight, namely, 150 mg/day, which was below at a minimum dose but was well tolerated at normal urinary oxalate values. Kidney transplantation performed in this patient has the potential to adversely affect pyridoxine acceptance.
 | Table 2. Pharmacotherapy of hypercalciuria in UEA on Guidelines for Urolithiasis (European Association of Urology, 2018 ). [Click here to view] |
 | Table 3. Use of pharmacotherapy in case reports of hyperoxaluria. [Click here to view] |
 | Table 4. Pharmacological treatment for hyperoxaluria. [Click here to view] |
Alkaline citrate
Alkaline citrate therapy options include sodium and potassium citrate used to prevent calcium oxalate stone formation and oral chemolitholysis. There are four cases attached to Table 3 with the use of alkaline citrate, both sodium and potassium citrate. Alkaline citrate is used in primary and secondary hyperoxaluria conditions. A diagnosis of primary hyperoxaluria with a mutation in the AGXT gene was confirmed in a 52-year-old patient after performing an allogeneic kidney transplant. A good prognosis was noted from taking vitamin B6 and potassium sodium hydrogen citrate orally in the waiting period so liver transplantation was not selected in this case. Another case of primary hyperoxaluria is attached to the table and another case of patients aged 10 years and 32 years also receiving potassium citrate. There are no specific data describing the outcome of using potassium citrate specifically in these cases, but the patient showed a good prognosis supported with a renal stone removal procedure. Potassium citrate was also used in patients with enteric/secondary hyperoxaluria in the case of an 8-year-old child in the table. Renal calculi in this patient were resolved within 1 year.
The UAE Guidelines of Urolithiasis and the England Journal of Medicine also recommend alkaline citrate in the treatment of primary hyperoxaluria, but the calculation of dose suitability is difficult to study because as a case it does not mention the dose of alkaline citrate or does not write down the patient’s weight. While the use of alkaline citrate in secondary hyperoxaluria is only recommended by the UAE Guidelines of Urolithiasis, the New England Journal of Medicine recommends calcium and magnesium supplements as agents to prevent the formation of calcium oxalate crystals.
Magnesium supplementation
The use of magnesium supplements has a similar function with alkaline citrate, an option for preventing calcium oxalate crystallization. Calcium supplements were used in one of the cases in the table. This case was a primary hyperoxaluria type 3 case in an elderly 78-year-old along with a hyperoxaluria treatment regimen. The patient had a history of kidney stones and chronic kidney disease, about 58 years, the etiology of the kidney stones experienced by the patient was not studied. When they worsened, they needed intervention and evaluation for 1 year until the diagnosis of primary hyperoxaluria type 3 was due to rare cases, so the intervention of pyridoxine therapy and others were ineffective when given and the patient’s prognosis reached renal failure, so hemodialysis was necessary. The use of magnesium supplements in primary hyperoxaluria is also recommended by the UAE Guidelines of Urolithiasis, but the calculation of dose suitability is difficult to study because as a case it does not mention the dose of use.
Calcium supplementation
The purpose of using calcium supplementation is to enable calcium oxalate complex formation in the intestine. Two cases reported the use of calcium supplements in the treatment regimen for hyperoxaluria with secondary hyperoxaluria. The first case of a patient with secondary hyperoxaluria (8 years) was cured with calcium supplements combined with hyperoxaluria medication, showing a good prognosis where 1 year after treatment there were no kidney stones and the hydronephrosis resolved. Another case was secondary hyperoxaluria in a 77-year-old patient. The outcome of using calcium supplements in the treatment of renal stones showed a good prognosis with decreased urinary oxalate level and the absence of kidney stone formation. The use of magnesium supplements in secondary hyperoxaluria is also recommended by the UAE Guidelines of Urolithiasis and the New England Journal of Medicine at a dose of 500–1,000 mg/day.
Other therapies
Cholestyramine and sodium bicarbonate are supportive therapies used in some cases. The use of cholestyramine is intended for the treatment of bile acid malabsorption, but colestipol and colesevelam should have similar effects and reduce urine oxalate. The dose of cholestyramine for hyperoxaluria therapy is 16 g daily in secondary hyperoxaluria (Asplin, 2016). Sodium bicarbonate is oral chemolitholysis based on urine alkalinization, improves urine pH, and treats hypocitraturia at a dose of 1.5 g, three times daily (European Association of Urology, 2018).
CONCLUSION
The imbalance of regulation of calcium and oxalate in the body leads to hypercalciuria and hyperoxaluria. The pathophysiology of hypercalciuria may be associated with an increase of calcium transport in the intestine, renal tubule reabsorption, and the regulation of bone mineral content, while the pathophysiology of hyperoxaluria includes AGT deficiency, GRHPR deficiency, HOGA defect, or increased intake of oxalic precursors. Reports on cases revealed that hypercalciuria was commonly treated with hydrochlorothiazide, chlorthalidone, and bendroflumethiazide, while hyperoxaluria was reduced by pyridoxine, alkaline citrate, magnesium supplementation, calcium supplementation, and other additional therapies.
ACKNOWLEDGMENTS
The authors would like to thank the Rector of Universitas Padjadjaran via the Directorate of Research and Community Engagement for facilitating the publication fee.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
AUTHOR CONTRIBUTIONS
Jutti Levita (JL) was principally responsible for the conception and design of the review and checked, finalized, and revised the manuscript. Lika Ginanti Febriana (LGF) searched and collected the articles and wrote the initial manuscript. Sri Adi Sumiwi (SAS) checked the collected articles. All authors read and approved the final manuscript to be published.
REFERENCES
Adnan WAHWM, Nigwekar SU. Nutritional and medical management of kidney stones. Nutr Med Manag Kidney Stones, 2019; 4:93–106; doi:10.1007/978-3-030-15534-6 CrossRef
Arrabal-Polo MA, Arias-Santiago S, Girón-Prieto MS, Abad-Menor F, Pintado FLC, Zuluaga-Gomez A, Arrabal-Martin M. Hypercalciuria, hyperoxaluria, and hypocitraturia screening from random urine samples in patients with calcium lithiasis. Urol Res, 2012; 40:511–5; doi:10.1007/s00240-012-0474-2 CrossRef
Asplin JR. The management of patients with enteric hyperoxaluria. Urolithiasis, 2016; 44:33–43; doi:10.1007/s00240-015-0846-5 CrossRef
Barlow W, Darrad M, Fletcher S, Devarajan R. Hypercalciuria: a rare contributing cause of exercise-induced visible haematuria. J Endoluminal Endourol, 2018; 1:e33–7; doi:10.22374/jeleu.v1i2.16 CrossRef
Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III — a model for studying perturbations in glyoxylate metabolism. J Mol Med, 2012; 2012:1497–504; doi:10.1007/s00109-012-0930-z CrossRef
Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol, 2015; 10:1257–72; doi:10.2215/CJN.09750913 CrossRef
Brzica H, Breljak D, Burckhardt BC, Burckhardt G, Sabolic I. Oxalate?: from the environment to kidney stones. Arch Ind Hyg Toxicol, 2013; 64:609–30; doi:10.2478/10004-1254-64-2013-2428 CrossRef
Burns Z, Knight J, Fargue S, Holmes R, Assimos D, Wood K. Future treatments for hyperoxaluria. Curr Opin Urol, 2020; 30:171–6; doi:10.1097/MOU.0000000000000709 CrossRef
Christakos S. Mechanism of action of 1, 25-dihydroxyvitamin D 3 on intestinal calcium absorption. Rev Endocr Metab Disord, 2012; 13:39–44; doi:10.1007/s11154-011-9197-x CrossRef
Chung H. The role of Randall plaques on kidney stone formation. Transl Androl Urol, 2014; 3:251–4; doi:10.3978/j.issn.2223-4683.2014.07.03
Cochat P, Rumsby G. Primary Hyperoxaluria. N Engl J Med, 2013; 369:649–58; doi:10.1056/NEJMra1301564 CrossRef
Curhan GC, Taylor EN. 24-H uric acid excretion and the risk of kidney stones. Kidney Int, 2008; 73:489–96; doi:10.1038/sj.ki.5002708 CrossRef
Curry JN, Yu ASL. Paracellular calcium transport in the proximal tubule and the formation of kidney stones. Am J Physiol—Ren Physiol, 2019; 316:F966–9; doi:10.1152/ajprenal.00519.2018 CrossRef
Dhayat NA, Faller N, Bonny O, Mohebbi N, Ritter A, Pellegrini L, Bedino G, Schönholzer C, Venzin RM, Hüsler C, Koneth I, Del Giorno R, Gabutti L, Amico P, Mayr M, Odermatt U, Buchkremer F, Ernandez T, Stoermann-Chopard C, Teta D, Rintelen F, Roumet M, Irincheeva I, Trelle S, Tamò L, Roth B, Vogt B, Fuster DG. Efficacy of standard and low dose hydrochlorothiazide in the recurrence prevention of calcium nephrolithiasis (NOSTONE trial): Protocol for a randomized double-blind placebo-controlled trial. BMC Nephrol, 2018; 19:1–9; doi:10.1186/s12882-018-1144-6 CrossRef
DiBianco JM, Jarrett TW, Mufarrij P. Metabolic syndrome and nephrolithiasis risk: should the medical management of nephrolithiasis include the treatment of metabolic syndrome? Rev Urol, 2015; 17:117–28; doi:10.3909/riu0650
Enr?quez R, Sirvent AE, Amoros F, Martinez M, Reyes JB, Cabezuelo A. Renal hypomagnesemia, hypercalciuria and nephrocalcinosis in a middle-aged man. Scand J Urol Nephrol, 2003; 37:93–5. CrossRef
Ermer T, Eckardt K, Aronson PS, Knauf F, Haven N. Oxalate, inflammasome, and progression of kidney disease Theresa. Curr Opin Nephrol Hypertens, 2017; 25:363–71; doi:10.1097/MNH.0000000000000229.Oxalate CrossRef
European Association of Urology. EAU guidelines on urolithiasis. European Association of Urology, London, UK, 2018.
Felsenfeld A, Rodriguez M, Levine B. New insights in regulation of calcium homeostasis. Curr Opin Nephrol Hypertens, 2013; 22:371–6; doi:10.1097/MNH.0b013e328362141e CrossRef
Figueres L, Hourmant M, Lemoine S. Understanding and managing hypercalciuria in adults with nephrolithiasis: keys for nephrologists. Nephrol Dial Transplant, 2020; 35:573–5; doi:10.1093/ndt/gfz099 CrossRef
Fisang C, Anding R, Müller SC, Latz S, Laube N. Urolithiasis — an interdisciplinary diagnostic, therapeutic and secondary preventive challenge. Dtsch Arztebl Int, 2015; 112:83–91; doi:10.3238/arztebl.2015.0083 CrossRef
Fontenelle LF, Sarti TD. Kidney stones: treatment and prevention. Am Fam Physician, 2019; 99:491–6.
Frick KK, Bushinsky DA. Molecular mechanisms of primary hypercalciuria. J Am Soc Nephrol, 2003; 14:1082–95; doi:10.1097/01.ASN.0000062960.26868.17 CrossRef
García Nieto VM, Luis-Yanes MI, Tejera-Carreño P, Pérez-Suarez G, Moraleda-Mesa T. The idiopathic hypercalciuria reviewed. Metabolic abnormality or disease? Nefrologia, 2019; 39:592–602; doi:10.1016/j.nefroe.2019.12.005 CrossRef
Glew RH, Sun Y, Horowitz BL, Konstantinov KN, Barry M, Fair JR, Massie L, Tzamaloukas AH. Nephropathy in dietary hyperoxaluria: a potentially preventable acute or chronic kidney disease. World J Nephrol, 2014; 3:122–42; doi:10.5527/wjn.v3.i4.122 CrossRef
Huang Z, Liu Y, Qi G, Brand D, Zheng S. Role of vitamin A in the immune system. J Clin Med, 2018; 7:258; doi:10.3390/jcm7090258 CrossRef
Ivanovski O, Drüeke TB. A new era in the treatment of calcium oxalate stones? Kidney Int, 2013; 83:998–1000; doi:10.1038/ki.2013.41 CrossRef
Jacobs TP, Kaufman M, Jones G, Kumar R, Schlingmann K, Shapses S, Bilezikian JP. A lifetime of hypercalcemia and hypercalciuria, finally explained. J Clin Endocrinol Metab, 2014; 99:708–12; doi:10.1210/jc.2013-3802 CrossRef
Keller J, Schinke T. The role of the gastrointestinal tract in calcium homeostasis and bone remodeling. Osteoporos Int, 2013; 24:2737–48; doi:10.1007/s00198-013-2335-4 CrossRef
Llanas B, Richard E, Blouin J. Late diagnosis of primary hyperoxaluria type III. Ann Clin Biochem, 2017; 54:406–11; doi:10.1177/0004563216677101 CrossRef
Martines DGL De, Gianotten S, Wetzels JFM, Meijden WAG Van Der. Secondary hyperoxaluria due to pancreatic insufficiency. Neth J Med, 2019; 77:287–92.
Mitchell XT, Kumar P, Reddy T, Wood KD, Knight J, Assimos DG, Holmes RP. Dietary oxalate and kidney stone formation. Am J Physiol Ren Physiol, 2021; 316:409–13; doi:10.1152/ajprenal.00373.2018 CrossRef
Narang G, Shimon T, Moore J, Hager M, Pinto F, Stern K, Keddis M, Humphreys M. Novel variant in CLDN16?: a further step in the diagnosis of familial hypomagnesemia with hypercalciuria and nephrocalcinosis — a case report. Uro, 2021; 2021:76–81. CrossRef
Nazzal L, Puri S, Goldfarb DS. Enteric hyperoxaluria: an important cause of end-stage kidney disease. Nephrol Dial Transplant, 2016; 31:375–82; doi:10.1093/ndt/gfv005 CrossRef
Pak CY, Sakhaee K, Moe OW, Poindexter J, Huet BA. Defining hypercalciuria in nephrolithiasis. Int Soc Nephrol, 2015; 80:777–82; doi:10.1038/ki.2011.227.Defining CrossRef
Pak CYC. Pharmacotherapy of kidney stones. Expert Opin Pharmacother, 2008; 9:1509–18; doi:10.1517/14656566.9.9.1509 CrossRef
Penido MGMG, Tavares M de D. Bone disease in pediatric idiopathic hypercalciuria. World J Nephrol, 2012; 1:54; doi:10.5527/wjn.v1.i2.54 CrossRef
Pu F, Chen N, Xue S. Calcium intake, calcium homeostasis and health. Food Sci Hum Wellness, 2016; 5:8–16; doi:10.1016/j.fshw.2016.01.001 CrossRef
Rahman N, Hitchcock R. Case report of paediatric oxalate urolithiasis and a review of enteric hyperoxaluria. J Pediatr Urol, 2010; 6:112–6; doi:10.1016/j.jpurol.2009.06.013 CrossRef
Richards AJ. Idiopathic hypercalciuria. Br Med J, 2006; 1:47; doi:10.1136/bmj.1.5739.47-b CrossRef
Riedel TJ, Knight J, Murray MS, Milliner DS, Holmes RP, Lowther WT. Biochimica et biophysica acta 4-hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. BBA—Mol Basis Dis, 2012; 1822:1544–52; doi:10.1016/j.bbadis.2012.06.014 CrossRef
Ryan LE, Ing SW. Idiopathic hypercalciuria?: can we prevent stones and protect bones?? Cleve Clin J Med, 2021; 85:47–54; doi:10.3949/ccjm.85a.16090 CrossRef
Ryan LE, Ing SW. Idiopathic hypercalciuria and bone health. Curr Osteoporos Rep, 2012; 25:286–95; doi:10.1007/s11914-012-0120-5 CrossRef
Siener R, Schade N, Claudia N, Von, GE, Unruh, Hesse A. The efficacy of dietary intervention on urinary risk factors for stone formation in recurrent calcium. J Urol, 2005; 173:1601–5; doi:10.1097/01.ju.0000154626.16349.d3 CrossRef
Singh P, Chebib FT, Cogal AG, Gavrilov DK, Harris PC, Lieske JC. Pyridoxine responsiveness in a type 1 primary hyperoxaluria patient with a rare (Atypical) AGXT gene mutation. Kidney Int Rep, 2020; 5:955–8; doi:10.1016/j.ekir.2020.04.004 CrossRef
Skalova S, Konrad M, Kutilek S. Three different causes of hypercalciuria. K Padiatr, 2011; 223:287–9. CrossRef
Sultana A, Kumar U, Hanif M. Primary hyperoxaluria: a case report and brief review. Paed Neph J Bang, 2020; 5:50–4.
Tasic V, Dervisov D, Koceva S, Weber S, Konrad M. Hypomagnesemia with hypercalciuria and nephrocalcinosis: case report and a family study. Pediatr Nephrol, 2005; 20:1003–6; doi:10.1007/s00467-005-1853-5 CrossRef
Vezzoli G, Soldati L, Teresa A, Terranegra A, Biasion R, Russo RC, Lauretani F, Bandinelli S, Bartali B, Cherubini A, Cusi D, Ferrucci L. Urinary calcium is a determinant of bone mineral density in elderly men participating in the InCHIANTI study. Kidney Int Vol, 2014; 67:2006–14. CrossRef
Witting C, Langman CB, Assimos D, Baum MA, Kausz A, Milliner D, Tasian G, Worcester E, Allain M, West M, Knauf F, Lieske JC. Pathophysiology and treatment of enteric hyperoxaluria. Am Soc Nephrol, 2021; 16:1–9; doi:10.2215/CJN.08000520 CrossRef
Worcester EM, Bergsland KJ, Gillen DL, Coe FL. Evidence for increased renal tubule and parathyroid gland sensitivity to serum calcium in human idiopathic hypercalciuria. Am J Physiol Ren Physiol, 2021; 305:853–60; doi:10.1152/ajprenal.00124.2013 CrossRef
Zerwekh JE. Bone disease and idiopathic hypercalciuria Joseph. Semin Nephrol, 2008; 23:1–7.
Zhao Y, Yang Y, Zhou P, Jiang J, Chen Z, Du D. Novel mutations in response to vitamin B6 in primary hyperoxaluria type 1 after only kidney transplantation: a case report. Transl Androl Urol, 2020; 9:2848–54; doi:10.21037/tau-20-979 CrossRef