
INTRODUCTION
Drug research entails determining the safety and toxicity of potential new drugs, as well as developing a target receptor hypothesis for a specific condition and screening the new drug candidates’ in vitro and/or in vivo biological activities. Conducting drug metabolism and pharmacokinetics tests, also known as ADMET, which means absorption, delivery, metabolism, elimination, and toxicity experiments, is an important part of drug discovery (Guoli et al., 2021; Leonardo and Adriano, 2019; Longfei et al., 2019; Yuhua et al., 2019). The use of early absorption, delivery, metabolism, and excretion (ADME) assessment has significantly reduced the number of compounds that fail clinical trials (Akbar et al., 2017; Kaitin, 2008; Mishra and Dahima, 2019).
Preclinical ADME’s main goal is to remove poor drug candidates early in the drug development process, allowing resources to be spent on promising candidates (Mishra and Dahima, 2019). Since the 1950s, regulatory agencies have focused on in vivo research to predict how new molecules would behave in the body of humans (Arora et al., 2008). Bioavailability, pharmacokinetics, metabolism, tissue distribution, and toxicity are usually evaluated in one non-rodent and one rodent species preceding administering a medication to a person in a clinical trial (dog or nonhuman primate) in phase 1. Radioactively labeled compounds are commonly used in the appropriate technique for biodistribution assessment. Animal studies and synthesizing enough radioactively labeled compounds both take time and resources (Oldendorf, 1970).
As a result, these assays are used later in the preclinical research process, when there are more tools available to study the few molecules that have made it that far. Thanks to advancements in cell and molecular biology, high-throughput sampling, and miniaturization technology in the 1990s, as well as stem cell-derived models at the turn of the century, early in silico ADME experiments have been developed to predict in vivo animal and human outcomes at a pace and cost-effectiveness suitable for the early discovery period (Mishra and Dahima, 2019; Oldendorf, 1970).
Preclinical drug testing necessitates the use of animals, which is time-consuming and expensive, as well as potentially causes individual pain. This has compelled scientists to look for ways to cut down on not just the time expended on drug detection experiments, but also the number of animals involved and their care by humans. To achieve this end, several new in silico techniques known as “alternatives” or “substitutes” for animal use in drug research have been developed (Arora et al., 2008).
The benefits of these choices involve a reduction in the number of animals utilized, the capability to deliver results rapidly, cost savings, and the versatility to monitor the experiment’s variables (Mishra and Dahima, 2019). The advancement of ADME profiling has reduced the number of drug candidates failing in clinical trials due to ADME issues, while also providing critical early information for drug candidate safety and toxicity prediction.
The SwissADME web tool is a free piece of software that predicts the following: physicochemical properties, distribution, metabolism, elimination, absorption, and also pharmacokinetic properties of molecules under investigation, all of which are important steps in the process of moving forward with clinical trials (Mishra and Dahima, 2019; Yusuf et al., 2020). Lipophilicity, flexibility, saturation, polarity, size, and solubility are among the six most significant physicochemical properties considered (Pires et al., 2018).
Parameters in druglikeliness can be investigated using a collection of rules developed by different pharmaceutical companies, which establish the correlation between biological activities and properties of pharmacokinetic, and that must be followed for in vivo action (Cheng et al., 2012). For instance, Pfizer established the Lipinski rule of five which states that the molecule’s molecular weight should be less than 500 Da, also the H-bond acceptor is less than 10, the H-bond donor should be higher than 5, the logP value should be less than 5, and biological transporters should be avoided (Mishra and Dahima, 2019).
The logP value must range from −0.4 to +5.6; the molar refractivity must be between 40 and 130; the molecular weight must be within the range of 180–480; and the number of the atoms must be between 20 and 70, including hydrogen bond donor and acceptor, according to Amgen’s Ghose rule (Cheng et al., 2012). The Egan rule notes that the logP value and topological polar surface area (TPSA) should not exceed 5.88 and 131.6, respectively, for predicting human intestinal absorption (Mishra and Dahima, 2019).
Based on small molecule lipophilicity and polarity computations, the BOILED-egg model is proposed as an efficient predictive model. The permeability glycoprotein (P-gp) substrate is used to measure active efflux across biological membranes. It defends the Central Nervous System (CNS) from xenobiotics and is overexpressed in tumor cells. Five main isoforms, namely CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, are substrates for 50%–90% of molecules. Inhibition of these isoenzymes is likely to be one of the more frequent sources of pharmacokinetics-related drug–drug interactions and can result in toxic ADME due to drug/metabolite accumulation (Potts and Guy, 1992).
TUG-770, 3-(4-((2-(cyanomethyl)phenyl)ethynyl)-2-fluorophenyl)propanoic acid, is a GPR-40 inhibitor in clinical trials (Fig. 1). This study aims to predict the in silico ADME experiments of TUG-770, to understand the predictable outcome of clinical trials, to find a link between in vivo and in silico outcomes, and to enhance TUG-770’s structure such that biological activity is not harmed while unwanted ADME effects are decreased. To our knowledge, this is the first research using SwissADME predictor to conduct in silico ADME studies of TUG-770 for the treatment of type 2 diabetes.
 | Figure 1. Chemical structure of TUG-770. [Click here to view] |
MATERIALS AND METHODS
SwissADME is a free online platform for determining the drug-likeness, pharmacokinetics, and medicinal chemistry related to the stability of small molecules (Noemi et al., 2020; Tianshu et al., 2021). In comparison to the state-of-the-art of ADME (free web-based tools) and pharmacokinetics, such as pk-CSM (Pires et al., 2018) and admetSAR (Cheng et al., 2012), and aside from unique access to proficient methods, such as ilogP (Daina et al., 2014) or the BOILED-egg (Daina and Zoete, 2016), strong points of SwissADME include input methods diverse, computation for various molecules, and the ability to display, save, and exchange results (per-molecule basis) or via global, and interactive graphs. SwissADME is considered now part of the SwissDrugDesign workspace (Daina et al., 2014; Daina and Zoete, 2017).
ChemDraw was used to draw the 2D structure of TUG-770, and ChemDraw 3D-Ultra version 19.0.0.22 was used to draw the 2D and 3D structures by minimizing energy with MM2 and MOPAC, with the minimum RMS gradient set to 0.01. The structure smiley was introduced after the structure was imported. The results of the SwissADME drug design study have been recorded.
RESULTS AND DISCUSSION
According to Verber, a GlaxoSmithKline (GSK) pharmaceuticals spokesperson, some drugs, such as steroids, should have a molecular weight of more than 500 Da, 10 or even lower rotatable bonds, and a polar surface area of lower than 140 Ao. The Muegge rule, proposed by Bayer Pharmaceuticals, defined the following parameters: number of rings (> 7), number of carbon atoms (< 4), number of heteroatoms (> 1), number of rotatable bonds (< 15), hydrogen bond donor atoms (< 5), and hydrogen bond acceptor (> 10). Molecular weight ranged from 200 to 600 D; logP was between the range −2 and +5; the topological surface area was < 150); and the F-value of Abbott bioavailability does not exceed 10% (Oldendorf, 1970).
For oral bioavailability and absorption, a drug must have strong aqueous solubility. Based on the latter, three methods, namely Estimated SOLubility (ESOL), A method to compute log S and to estimate the water solubility (ALI) logS, and (SILICOS-IT) logS (Vázquez-Tato et al., 2021), are used to estimate the water solubility. The ESOL model is an acronym for calculating aqueous solubility straight from chemical molecular structure, then through molecular weight, the heavy atoms proportion found in the aromatic system, and rotatable bonds number. The model predicted solubility correctly within a factor of 5–8 through three validation sets (Arora et al., 2008; Cheng et al., 2012). The in-silico prediction model of aqueous solubility utilizing logS (ALI) considers the effect of TPSA. By using a fragmental form, Log (SILICOS-IT) calculates the negative sign logarithm of a compound’s water solubility. The logS scale ranges from −10 (insoluble), −6 (poorly soluble), −4 (soluble), −2 (extremely soluble), and 0 (highly soluble) (Mishra and Dahima, 2019).
A drug’s lipophilic character must be strong enough to allow it to cross the cell membrane and have good biological activity. Several methods can be used to evaluate the lipophilicity parameter. The ilogP (in-house physics-based approach) is based on solvation-free energies using n-octanol and water measured using the model of a generalized born and solvent accessible surface area. On the other hand, XlogP3 is an atomistic approach that uses the XlogP software to measure corrective factors and a knowledge-based library. WlogP is a fragmental atomistic process. MlogP is a topological approach that uses 13 molecular descriptors in a linear relationship. a Belgian consultancy company that was founded in 2010 and that is specialized in computational drug design (SILICOS-IT) logP (hybrid method) uses fragment and topological descriptors; logP is a hybrid method. The arithmetic mean of the five lipophilic character predictions is the consensus of logP (Cheng et al., 2012).
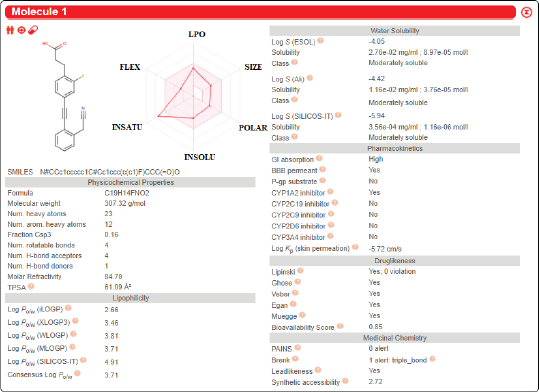
Figure 2 shows that the radar of bioavailability, revealed by the colored zone, is the best physicochemical space indicator for oral bioavailability when lipophilicity, saturation, size, flexibility, polarity, and solubility are considered. LogP can have lipophilicity of −0.7 to +5.0. The molecular weight can be anywhere between 150 and 500 (g/mol). Between 20 and 130 Ao is the TPSA (Antoine et al., 2017). The logS (ESOL) insolubility ranges from 0 to 6. The number of rotatable bonds should range from 0 to 9 and the unsaturation fraction ranges from 0.25 to 1.0, indicating that the carbon atom fraction in sp3 hybridization cannot be less than 0.25.
According to its physicochemical properties, the compounds’ molecular formula is C19H14FNO2. The molecular mass was 307.32 (g/mol). This complex comprises 23 heavy atoms and 12 aromatic heavy atoms. 0.16% of carbon atoms are active in the sp3 hybridization. There are four rotatable bonds, four hydrogen bond acceptors, and one H-bond donor in this structure. The molar refractivity was found to be 84.70, and the TPSA was measured to be 61.09 Ao (Mishra and Dahima, 2019). The logPo/w (ilogP), logPo/w (XlogP3), logPo/w (WlogP), logPo/w (MlogP), Po/w (SILICOS-IT), and consensus logPo/w were 2.66, 3.64, 3.81, 3.71, 4.91, 3.71, respectively. The logP values mean that the compound is lipophilic in general. With a logS (ESOL) value of −4.05, the compound’s water solubility was measured, meaning that it is moderately water-soluble.
The BOILED-egg model was used to study the pharmacokinetic properties. The BOILED-egg model helps in performing the computation of derivative polarity as well as lipophilicity since it generates datasets with high precision, speed, and transparency (Fahmina et al., 2020; Sabitu et al., 2020). Also, it aids in drug production by allowing chemical libraries to be purified. This model helps for intuitive estimation of passive gastrointestinal absorption (HIA) and brain penetration (BBB) concerning the direction of the molecules (Fig. 3). The yolk (yellow region) represents a high likelihood of brain invasion, while the white area (intestinal tract) represents a high likelihood of passive absorption by the GI tract. It is not appropriate for white and yolk areas to be mutually exclusive.
TUG-770 has a high degree of gastrointestinal absorption and easily passes through the blood–brain barrier. There will be no problems with opioid excretion since there is no P-gp substrate. P-gp is necessary for drug removal and absorption. P-gp has a stronger impact on limiting drug absorption from blood circulation into the brain and from the intestinal lumen into epithelial cells due to its localization than it does on facilitating drug excretion from hepatocytes and renal tubules into the neighboring luminal space due to its localization. Since it activates the isoenzymes CYP2C19 and CYP2D6, there is a risk of aggregation or drug–drug reactions, which could lead to toxicity.
 | Figure 2. The bioavailability radar of TUG-770 using SwissADME predictor. [Click here to view] |
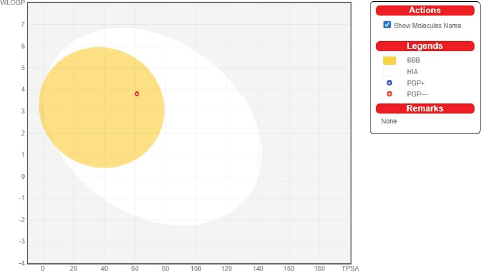 | Figure 3. The presence of a molecule in the yolk of an egg indicates that it would be able to cross the blood–brain barrier. [Click here to view] |
With a bioavailability score of 0.85, the parameter of drug-likeness is considered high, since it fits the Lipinski, Verber, and Egan rule. The score of Synthetic Accessibility of SwissADME is based on the premise that the occurrence of molecular fragments in “really” obtainable molecules is related to synthesis ease. For typical chemical moieties, the fragmental contribution to Synthetic Accessibility should be beneficial, but for uncommon moieties, it should be unfavorable. The synthetic usability score was discovered to be 2.72, suggesting that the molecule is not difficult to synthesize. There is no alarm about Pan-Assay Interference Compounds (PAINS), suggesting that it is a very particular compound.
OUTPUT
We used its incorporation into SwissADME to improve the graphical efficiency by predicting P-gp substrates, which is the most effective active efflux mechanism found in biological barriers. The state of the computation is shown immediately after application, beginning with validation of structure. This transient page communicates with the workload management system (Slurm version 17.11.2) to inform the user that his or her job has been queued or is being worked on. The consumer will see the various stages of the computation and observe the overall procedure on a progress bar. Starting calculations take between 15 and 20 seconds for a compound the size of a druglike molecule. Predictions are shown on the first performance page until the progression bar is filled (Antoine et al., 2017).
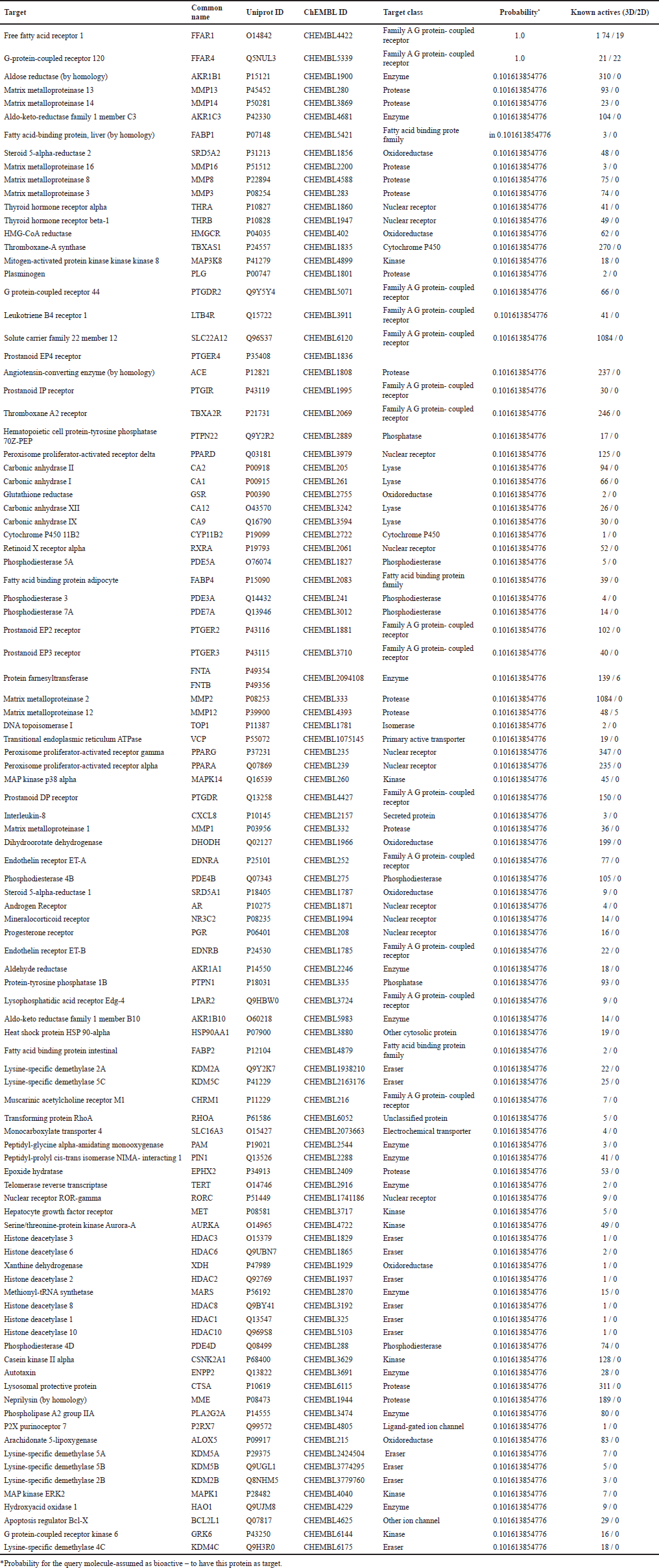 | Table 1. SwissTargetPrediction. [Click here to view] |
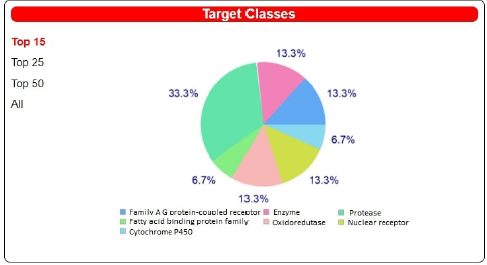 | Figure 4. Page showing the outcome of the prediction. The projected targets for the question molecule are shown on this page. Like their scores, targets are ranked. The distribution of goal groups is shown in a pie chart which is used to denote predictions dependent on homology. [Click here to view] |
The most valuable piece of information on this result page is the tabulated list of potential protein targets (Table 1), as determined by the dual-score ligand-dependent reverse screening of the query molecule against the collection of known actives. The table rows are ranked by default based on the likelihood of the related protein being the real target of the query molecule. Protein complex targets are now provided incomplete names (linked to a particular ChEMBL ID), with their subunits/components linked to Genecard and Uniprot separately in version 2019.
The probability values, shown as green bars, are calculated using the combined scores of the most closely related compounds to the query molecule (in 2D and 3D) that are active on a given protein as previously stated, and outlined in detail elsewhere (Antoine et al., 2019; Gfeller et al., 2014). Importantly, this value accurately depicts the likelihood of a bioactive molecule having a specific protein as a goal, but not the likelihood of being bioactive. Furthermore, targets labeled with the phrase “by homology” are projections focused on related molecules active on proteins with a high degree of homology (Antoine et al., 2017).
A common example is an orthology in which a target is expected for a given species depending on the question molecule’s similarities to compounds active on ortholog proteins. In the last column, you can see the number of compounds that are active on each listed target and are extremely like the query molecule. You may adjust the number of planned targets shown on a website to 15 (default), 25, 50, or all using page scanning (maximum 100). Furthermore, by clicking on the header, the table can be arranged by any column, and the results can be filtered using a search box. Furthermore, users can use advanced export options by clicking on dedicated icons. The table can be saved in a variety of formats such as CSV, Excel, and PDF, copied or printed straight from the browser. A pie chart created with JPgraph is shown in a box located on the top-right of the result sheet (Fig. 4) as a description of the forecast target groups (column 5 of Table 1) (version 4.2.6). The percentages equate to the top 15 proteins by design, but the user can change this by selecting the top 25, 50, or all expected targets using the buttons on the left of the column. By clicking on the pie-chart, a full-size image would appear in a new tab of the window, allowing for more saving or printing. The user’s question molecule’s chemical composition is shown in a box located on the top-left side of the result sheet (Fig. 4). the molecular modeling group of the SIB Swiss Institute of Bioinformatics has created four icons that appear within this box and allow for the clear submission of the molecule of interest to other web resources provided. These interoperability symbols can be used in all boxes referring to the chemical compositions of known actives, as well as a reference to the ChEMBL Compound Report Card.
The above is classified in order of their similarity to the question molecules, from most to least. The “twins” icon sends the molecule to SwissSimilarity for ligand-based direct screening (Daina and Zoete, 2017), the “target” icon resubmits the aim prediction (on a different animal, for example), and the “pill” icon uses SwissADME to approximate physicochemical, ADME, or pharmacokinetic parameters (Zoete et al., 2016). The proposed expansion of interoperability by adding other in-house software is also in the works, so more icons will be added to the various websites in the future (Antoine et al., 2017). Obtaining the molecule’s SMILES through the fourth symbol also makes it easier to use it as a possible input to other, potentially external, processes. The ChEMBL ID, SMILES, similarity attribute to the query molecule, and, if applicable, the source species of the experimentally identified homologous target are all included in the file (Antoine et al., 2017).
Figure 4 shows a brief of the various target groups present among the projected targets in a pie chart. The chance calculated from the goal scores is shown as a horizontal bar in the fifth column. Ligands are sorted by how close they are to the question molecule. The ligands have a relation to their ChEMBL entries, and the resemblance to the question molecule is suggested. We propose that you explore the ligands manually that are closest and the query molecule to see how accurate the projections are, and which types of ligands have the most similarities to the query molecule. Finally, there are support pages along with interactive screenshots of the website, a FAQ page to help users, and a download page to get a little of the raw data used in the predictions process.
CONCLUSION
Traditional clinical trial methods require a significant expenditure in time and resources, and it is possible that the molecule will fail. Thus, to minimize or change the structure, in silico trials can be pursued rather than aggressively pursuing monopoly and running to animal studies. The SwissADME web tool allows users to calculate vital pharmacokinetic, physicochemical, and similar parameters for one or more molecules. It was inferred from the analysis that the compound’s aqueous solubility, as well as the sp3 hybridized carbon atoms fraction, should be increased. Further changes to the lead structure are needed to guarantee that the molecule does not hinder metabolizing enzymes.
SwissTargetPrediction is considered part of the Swiss Institute of Bioinformatics’ valuable initiative to offer online resources for computer-assisted drug design. SwissTargetPrediction can be combined further with these methods in the future, for example, by forecasting possible linking modes with Swiss-Dock (Gabriela and Walter, 2019; Gfeller et al., 2014).
TUG-770 has a high degree gastrointestinal absorption rate and quickly passes the blood-brain barrier. Because there is no P-gp substrate, there will be no issues with opioid excretion. Also, with its bioavailability score of 0.85, the drug-likeness parameter is considered high, since it meets the Lipinski, Verber, and Egan rules. Additionally, TUG-770 synthetic usability score was determined to be 2.72, indicating that the molecule is not difficult to synthesis. Moreover, the logP values indicate that the compound is lipophilic in general. The compound’s water solubility was determined with a logS (ESOL) value of −4.05, indicating that it is moderately water-soluble.
ACKNOWLEDGMENT
Declared None declared.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
AUTHORS’ CONTRIBUTIONS
All authors made significant contributions to the conception and design, acquisition of data, and analysis and interpretation of data. Also, they took part in writing the draft of the article and revising it in a better way; and agreed to submit to the current journal. They agreed about the final approval of the version to be published; and agreed to be accountable for all aspects of the work.
REFERENCES
Akbar A, Mohamed EIB, Raza S, Wajid R, Yeldez ELK, El Sayed ElAS, Nawaz T. Synthesis, characterization and in-silico ADMET screening of mono- and dicarbmethoxylated 6,6’-methylenebis(2-cyclohexyl-4-methylphenol) and their hydrazides and hydrazones. Der Chem Sin, 2017; 8:446−60.
Antoine D, Olivier M, Vincent Z. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1−13. CrossRef
Antoine D, Olivier M, Vincent Z. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res, 2019; 47:W357–64. CrossRef
Arora T, Mehta AM, Sharma KK. Substitute of animals in drug research: an approach towards fulfillment of 4R’s. Indian J Pharm Sci, 2008; 73:1−6.
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Info Model, 2012; 52:3099−105. CrossRef
Daina A, Michielin O, Zoete V. iLOGP: a simple, robust and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Info Model, 2014; 54:3284−301. CrossRef
Daina A, Zoete V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem, 2016; 11:1117–21. CrossRef
Daina A, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1−13. CrossRef
Fahmina Z, Anjali G, Karthick T, Kavita K, Ali AS, Anujit G, Poonam T, Nahid N. Physicochemical and pharmacokinetic analysis of anacardic acid derivatives. ACS Omega, 2020; 5:6021−30. CrossRef
Gabriela BF, Walter FAJ. Docking with SwissDock. Meth Mol Biol, 2019; 2053:189−202. CrossRef
Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res, 2014; 42:W32–8. CrossRef
Guoli X, Zhenxing W, Jiacai Y, Li F, Zhijiang Y, Changyu H, Mingzhu Y, Xiangxiang Z, Chengkun W, Aiping L, Xiang C, Tingjun H, Dongsheng C. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res, 2021; 49:W5–14. CrossRef
Kaitin KI. Obstacles and opportunities in new drug development. Clin Pharmacol Ther, 2008; 83:210−2. CrossRef
Leonardo LGF, Adriano DA. ADMET modeling approaches in drug discovery. Drug Discov, 2019; 24:1157−65. CrossRef
Longfei G, Hongbin Y, Yingchun C, Lixia S, Peiwen D, Weihua L, Guixia L, Yun T. ADMET-score—a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm, 2019; 10:148–57. CrossRef
Mishra S, Dahima R. In-vitro ADME studies of TUG-891, a GPR-120 inhibitor using SwissADME predictor. J Drug Deliv Ther, 2019; 9:266−369.
Noemi ADI, Bárbara IDE, José LMF. In silico ADME/Tox profiling of natural products: a focus on BIOFACQUIM. ACS Omega, 2020; 5:16076–84. CrossRef
Oldendorf WH. Measurement of brain uptake of radiolabeled substances using a tritiated water internal standard. Brain Res, 1970; 24:372−6. CrossRef
Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem, 2018; 58:4066−72. CrossRef
Potts RO, Guy RH. Predicting skin permeability. Pharm Res, 1992; 9:663–9. CrossRef
Sabitu BO, Adamu U, Gideon S, Sani U. QSAR modeling, molecular docking and ADMET/pharmacokinetic studies: a chemometrics approach to search for novel inhibitors of norepinephrine transporter as potent antipsychotic drugs. J Iran Chem Soc, 2020; 17:1953–66. CrossRef
Tianshu Y, Yunkai Y, Yan W. Predictive biomarkers and potential drug combinations of epi-drugs in cancer therapy. Clin Epigenetics, 2021; 13:113−31. CrossRef
Vázquez-Tato MP, Julio AS, Francisco M, Francisco F, Santiago de F, Javier M, Juan VT, Aida J, Victor HS, José VT. Highly hydrophilic and lipophilic derivatives of bile salts. Int J Mol Sci, 2021; 22:6684−98. CrossRef
Yuhua L, Qiang M, Mengbi Y, Dongyang L, Xiangyu H, Lan T, Xin W, Yuanfeng L, Xiaoyan C, Kexin L, Ai-Ming Y, Zhong Z, Huichang B. Current trends in drug metabolism and pharmacokinetics. Acta Pharm Sin B, 2019; 6:1113−44. CrossRef
Yusuf I, Adamu U, Sani U. Computational studies of a series of 2-substituted phenyl-2-oxo-, 2-hydrox-yl- and 2-acylloxyethylsulfonamides as potent anti-fungal agents. Heliyon, 2020; 6:1−6. CrossRef
Zoete V, Daina A, Bovigny C, Michielin O. SwissSimilarity: a web tool for low to ultra-high throughput ligand-based virtual screening. J Chem Inform Comput Sci, 2016; 56:1399–404. CrossRef