
INTRODUCTION
Voluminous data of the literature illustrated that the diet ingredients of fruit, vegetables, and plant parts having capabilities to manage hypercholesterolemia by the existing potent bioactive phytocompounds such as polyphenol, terpenoids, flavonoids, and alkaloids (Rastogi et al., 2016). In a similar context, caffeic acid is a metabolite of hydroxycinnamate of the non-flavonoids phenolic acid mostly obtained from edible fruits, vegetables, herbs, and stress climatic plants which are used in therapeutics of numerous ailments. This also exhibits numerous processed dietary sources because of antioxidant properties due to their free radical scavenging capabilities as well as potent ligand can interact with several metabolic targeted protein and enzymes (Agunloye and Oboh, 2018; Laranjinha and Cadenas, 1999). In plant metabolism, caffeic acid or caffeic acid-like compounds furnish a series of hydroxycinnamic acids (C6–C3) that vary from an individual by the quantity of hydroxy and methoxy groups on their phenyl unit (Park, 2009). These kinds of monophenol carboxylic acids are repeatedly found to be esterified to polyols. Accordingly, 3,4-dihydroxycinnamic acid (caffeic acid) is combatted in average-sized polyester branches of the tetraolicquinic acid, i.e., 3,5-di-O-caffeoylquinic acid, that occurs in coffee beans. Along with this, there are numerous reports on the biomedicinal uses, such as antidiabetic, antioxidants, antihyperlipidemic, neuroprotective, anti-inflammatory, antioncogenic, and endocrinal booster, of caffeic acid (de Alencar Silva et al., 2020; Tsai et al., 2011). Consequently, the derivatives of caffeic acid also have significant therapeutic applications. Besides this, the active pharmaceutical agent needed suitable bioavailability in accordance with the five rules of the Lipinski (Koehn and Carter, 2005; Kumar, 2016). Thus, most of the polyphenols are considered active pharmaceutical agents because of their ideal profile of the bioavailability and capability to cross the blood–brain barrier (BBB). Therefore, the design of ideal polyphenol-based or polyphenol-stirred drugs against the protein targets may appropriately be a promising direction for future research on polyphenols. Based on the above fact, the current study was planned to investigate the HMG-CoA reductase (HMGCR) inhibition capabilities of caffeic acid by using in-vitro and in-silico examinations.
MATERIAL AND METHODS
In vitro HMGCR inhibition assay
The HMGCR inhibition activity of the caffeic acid was assessed by using the commercially available assay kit (Sigma-Aldrich) based on spectrophotometric measurement through 96 well plate (Liang et al., 2015; Saeedi Saravi et al., 2017). The HMGCR concentration was taken as a stock solution (0.50–0.70 mg/ml). The stock solution of caffeic acid compound was prepared at 0.02 mg/ml. The 1 μl volumes of the test samples were mixed with the reaction mixture containing 184 μl assay buffer, Nicotinamide Adenine Dinucleotide Phosphate (NADPH) (4 μl), HMG-CoA substrate (12 μl), and HMGR (2 μl). Pravastatin (Sigma Aldrich co.) and distilled water were used as the positive and negative controls, respectively. The IC50 values of caffeic acid and pravastatin were calculated by the online calculator of AAT Bioquest (Anwar et al., 2020).
Inhibition kinetics of HMGCR
The inhibition kinetics of caffeic acid against HMGCR was determined by following standard method (Sjögren et al., 2009; Wang et al., 2015). A particular quantity of HMGCR was incubated with increasing concentrations of the caffeic acid at an optimum temperature of 37°C for 15 minutes, in the absence or presence of samples (concentration alike to IC50). Reactions were completed and absorption was estimated and transformed to the reaction by the Lineweaver–Burk plot.
Ligand preparation
The three-dimensional conformation chemical structure of caffeic acid was downloaded from PubChem (pubchem.ncbi.nlm.nih.gov) web interface of the National Institute of Health. The PubChem CID of the caffeic acid was 689043. The compound was further processed by using PyMol and Autodock tools for the analysis of molecular docking (Madhavi Sastry et al., 2013).
Target protein preparation
The molecular three-dimensional structure of the HMGCR protein [Protein Data Bank (PDB) ID 1DQA] was retrieved from a Protein Data Bank (www.rcsb.org) in PDB format. Pre-processing of protein was done by deleting the water molecules, and the addition of hydrogen atoms and further processes were performed with the help of Autodock tools (Madhavi Sastry et al., 2013; Rizvi et al., 2013).
Molecular docking
Molecular docking was performed by using the Autodock to investigate the interaction between the target protein (HMGCR) and ligands (Caffeic acid and pravastatin) (Iheagwam et al., 2019; Rizvi et al., 2013). The molecular docking was carried out based on ligand binding to a receptor protein using scoring functions. The default grid dimension predicted by the Autodock tools depending on the pocket region of the protein was considered for further analysis. As a result of molecular docking, binding energy in kcal/mol was generated. The molecular interaction of the protein and ligand in three dimensions was analyzsed by using the PyMol. The number of hydrogen bonds, bond length, and the interacting residues of the protein and ligand were acquired from Lig plot.
Molecular dynamics (MD) simulation
Caffeic acid was obtained for the MD simulation in a complex with the enzyme HMGCR. Simulation studies were carried out with Biovia discovery studio client 2020 by following the standard protocol (Brooks et al., 2021; Noha et al., 2017). Protein was prepared and any prebound ligand or unnecessary molecules were removed. Any missing stretch or residues were prefixed. The structure of the ligand i.e., caffeic acid was retrieved from Pubchem database as mentioned earlier. The protein–ligand complex was put in a solvated environment of water. CHARMM 36 forcefield was used to detect the constraints, allowing the MD simulation to run through an environment of periodic boundary to avoid unnecessary surface artifacts.
The solvated system was then put through 1,000 steps of steepest descent minimization and 1,000 steps of conjugate gradient minimization using the forcefield. Next, the entire system was then heated at 60 to 300 K in 6 ps, for a runtime of 200 ps. The initial confirmation was taken as a reference to determine the root mean score fluctuation (RMSF) of the entire complex.
ADMET assessment
ADMET analysis was carried out using Swiss ADME (https://www.swissadme.ch) web tool predictor to evaluate the absorption, distribution, metabolism, and excretion (ADME) properties of the compound. The access of physicochemical properties, lipophilicity, water solubility, pharmacokinetics profile, drug likeness, and medicinal chemistry properties of the phytocompounds can be predicted for the development of drug (Daina et al., 2017). Along with that the interpretation of iLOGP and Bioavailability Radar can also be done by using Swiss ADME. The evaluation of caffeic acid can be done based on their ability to pass the BBB and follow the Lipinski rule of five. Lipinski’s “Rule of Five” interprets whether the compound is orally well absorbed or not as a drug molecule. The bioavailability of the drug molecule can be predicted based on total polar surface area (TPSA) as per Weber’s rule. TPSA ≤ 140 Å indicates good oral bioavailability. An ideal compound following these criteria would be able to pass the BBB if the number of hydrogen bonds is between 8 and 10 and no acidic groups are present in the molecule.
Gastrointestinal absorption—a Brain Or IntestinaL EstimateD (BOILED-Egg) model
The BOILED-Egg permeation method is a predictive model for the estimation of two pharmacokinetic behavior, i.e., gastrointestinal passive absorption and the permeability of the blood–brain penetration barrier. It works with speed, accuracy, and conceptual ease by computing the polarity and lipophilicity of chemicals and generates clear graphical outputs (Daina and Zoete, 2016). Along with the pharmacokinetics, bioavailability also plays a major role in drug development.
Toxicity prediction
ProTox-?? (https://tox-new.charite.de/protox_??) web server is computational freely available in silico toxicity prediction tool consisting of 33 models to design the development of drug process. It incorporates the prediction of oral toxicities of chemicals which can be measured in different levels of toxicity mainly qualitatively in terms of endpoints as binary output (active or inactive) along with confidence score such as acute toxicity, hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, adverse outcomes (Tox21) pathways, and toxicity (LD50) values in mg/kg body weight. Toxicity classes (Class I–VI) can also be predicted based on their toxic doses. Prediction accuracy, average similarity along with LD50, and toxicity class were generated instantly for the prediction of acute toxicity and toxicity targets.
Statistical analysis
Data of the in-vitro determinations were performed through devoted AAT Bioquest, Inc. “Quest Graph™ IC50 Calculator.” Retrieved from https://www.aatbio.com/tools/ic50-calculator by following the standard statistical approaches of enzymatic kinetics.
RESULTS
In-vitro assay
The in-vitro assay performed inhibition by the increasing concentrations of caffeic acid and the target enzyme of the HMGCR at optimum temperature and specific time intervals which showed 83.29% and 85.83%, respectively. The IC50 values of caffeic acid and pravastatin were calculated by using the online calculator of AAT Bioquest shown 40.6 nM and 10.162 μM, respectively (Fig. 1A and B).
Inhibition kinetics of enzyme assay
The values of Km and Vmax of the caffeic acid were found to be 0.360198 ± 0.04251 (11.8%) and 91.0863 ± 1.65 (1.811%), respectively. The values of Km and Vmax of test compound were further compared with the positive control i.e., Pravastatin had Km value as 10.325 ± 0.9372 (9.077%) and Vmax value as 94.2661 ± 2.458 (2.607%) (Fig. 2A and B).
Molecular docking
Molecular docking result analysis of binding interactions of the ligand with HMG CoA reductase docked against PDB 1DQA in comparison to standard drug. As a result of Autodock Vina, the binding energy of the caffeic acid compound was found to be −6.9 kcal/mol. Further processing of the protein and ligand was done by Lig plot to find the number of hydrogen bonds, bond length, and interacting residues (Table 1). The interacting residues Arg641, Ser794, Ala639, and Asn642 were present in the catalytic groove of the protein (Fig. 3A and B).
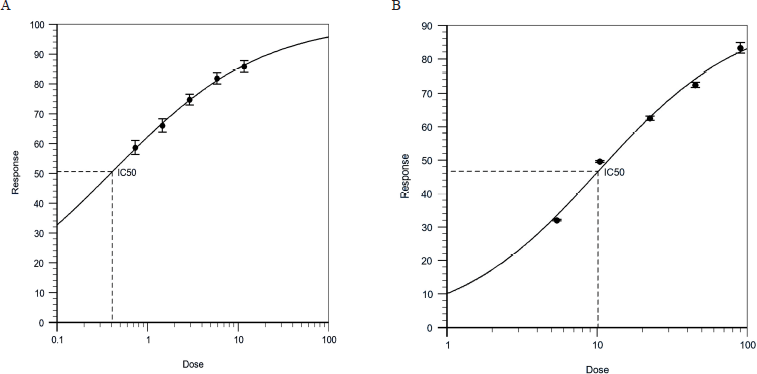 | Figure 1. A) In-vitro inhibitory activity of Pravastatin as positive control against HMGCR enzyme. (IC50 = 40.6 nM). B) In-vitro inhibitory activity of caffeic acid compound against HMGCR enzyme. (IC50 = 10.162 μM). [Click here to view] |
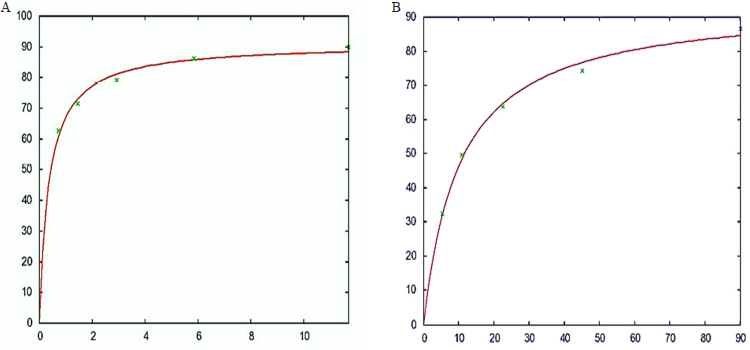 | Figure 2. A) Km and Vmax of Pravastatin as positive control against HMGCR enzyme was found to be 10.325 ± 0.9372 (9.077%) and 94.2661 ± 2.458 (2.607%) respectively. B) Km and Vmax of caffeic acid compound against HMGCR enzyme was found to be 0.360198 ± 0.04251 (11.8%) and 91.0863 ± 1.65 (1.811%) respectively. [Click here to view] |
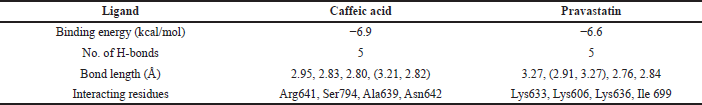 | Table 1. Molecular interaction studies of the caffeic acid against the target enzyme (HMG CoA reductase) [Click here to view] |
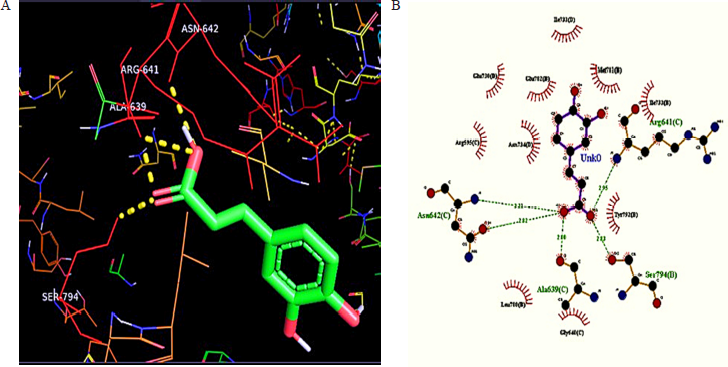 | Figure 3. A) Molecular interaction studies of ligand caffeic acid against HMGCR by docking analysis. B) Ligplot demonstrating the interactions of ligand-protein. Diagramsinterpret as follows: the residues involved in ligand bonds shown in thick lines (purple), residues involved in hydrogen binding with the ligand (black) with thin orange bonds along with their hydrogen bonds with the length in middle (olive green), and residues involved in non-bonded interactions or hydrophobic contacts between protein and ligand (red spikes). [Click here to view] |
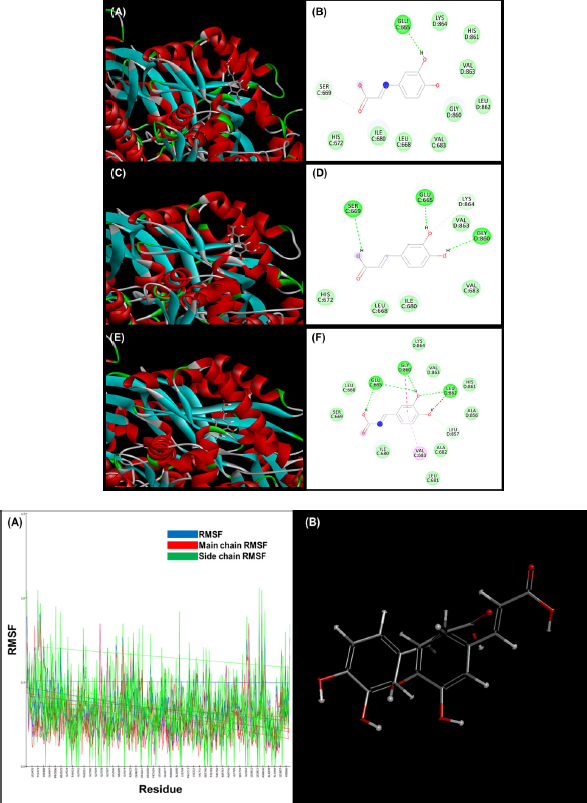 | Figure 4. A) MD simulation of caffeic acid with HMGCR over a period of 200ps showing no unwanted projection overall. (A,B) Screenshot and interactive residues at 60 ps. (C, D) Screenshot and interactive residues at 120 ps.(E, F) Screenshot and interactive residues at 200 ps. B) RMSF value along with of main chain and side chain RMSF values with respect to the amino acid residues of protein. (B)Molecular overlay of initial and final conformation after the simulation. [Click here to view] |
MDs simulation
MD simulation was performed using HMGCR -caffeic acid complex for around 200 ps (Fig. 4). The temperature constraints up to 300 K denotes a stable character of the complex. The potential energy was calculated on average which was found out to be, −277,696 kJ/mol. As shown in Figure 4A, RMSF values for the main chain and for the side chain that came out to be 1.41958 Å. This indicates the active binding of the compound to the protein structure. Even more the motion of the conformation was detected using biovia discovery studio client 2020 visualizer, the result shows a prompt interaction in a stable manner in all three snapshots. Even the molecular structure overlay also shows to overlap the presimulated and postsimulated conformations portraying no such alteration of position from the active cavity (Fig. 4B).
ADMET pharmacokinetics predictions
Pharmacokinetic (ADMET) profile of the caffeic acid compound was shown in table compared with the positive control such as pravastatin indicated an ideal drug profile. Further confirming the Lipinski rule of five as no violations has been shown by the compound indicating drug-likeness (Table 2). TPSA of the compound was found as 77.76 which characterize significant cellular plasma membrane permeability.
BOILED-Egg predictions
With the help of graphical classification model, the Egan’s BOILED-Egg permeation predictive model diagram has shown that the caffeic acid compound has good potential to cross Human Intestinal Absorption (HIA) (the passive human gastrointestinal absorption) but no BBB permeability. The compound was predicted as non-substrate of the p-glycoprotein (Fig. 5).
Toxicity
The prediction of toxicity of caffeic acid compound based on various properties, such as hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, LD50, and toxicity. The compound belonged to class 4; LD50 was predicted as 2,000 in mg/kg and was inactive as a toxicant but showed immunotoxicity (Table 3).
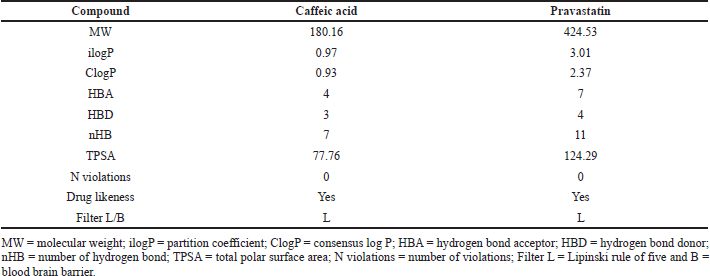 | Table 2. Physiochemical ADMET analysis of the caffeic acid against the Lipinski rule of five and BBB filter by Swiss ADMET web tool. [Click here to view] |
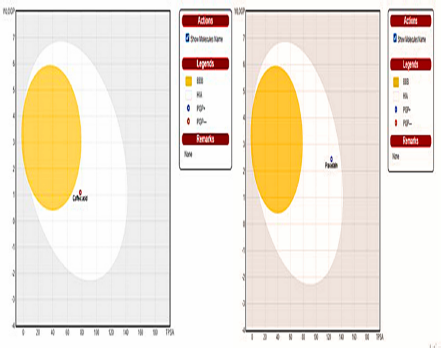 | Figure 5. Predicted BOILED-Egg model of caffeic acid and Pravastatin using SwissADME. [Click here to view] |
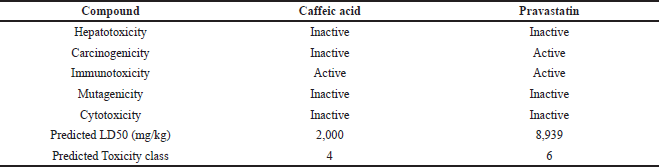 | Table 3. In-silico toxicity prediction of the caffeic acid by ProTox-II server. [Click here to view] |
DISCUSSION
Enzyme inhibitors, analogs, and inactivators include coarsely half of all the prescribed medicines which are using for health concerns (Holdgate et al., 2017). In the 1980s, statins, represented by atorvastatin and lovastatin, developed as inhibitors of 3?hydroxy?3?glutaryl CoA reductase (HMG-CoA reductase or HMGCR) and nowadays it is most prescribing drugs to treat hypercholesterolaemia (Marahatha et al., 2021; McKenney, 2003). Accordingly, it is a well-established fact that phytocompounds have capabilities to ameliorate numerous metabolic disorders by the interactions with particular target protein or enzymes, scavenging of free radicals, and other accelerative activities (Ademosun et al., 2015; Janice et al., 2020). In a similar context, caffeic acid is one of the leading ingredients of edible plant materials and dietary content which contributing to ameliorate numerous metabolic disorders as well as benefiting to the health (de Alencar Silva et al., 2020; Mukherjee et al., 2015; Tsai et al., 2011). The current study revealed that caffeic acid has a significant concentration-dependent HMGCR inhibition capability which relies on the strength of their bond to the enzyme. This kind of inhibition depicted the base structures of the compounds determine how well the molecule fits into the binding pocket of the target enzyme and binds with it. Whereas the pravastatin-like statin is synthetic statin which has an additional fluorinated phenol group and other moieties in the base structure that provide additional sites for binding within the enzyme pocket (Salvamani et al., 2016). Therefore, the test compound i.e., caffeic acid and pravastatin have been shown different inhibition capabilities as well as different IC50 in this study. The IC50 value of the test compound against the enzyme depending upon the concentrations of the target molecule, the inhibitor, and the substrate or ligand along with chemical and physical experimental milieus (Srinivas, 2016; Zeytünlüo?lu and Zihnio?lu, 2015). Similarly, the significant reaction rates shown by the values of Km and Vmax of the in-vitro assays of HMGCR inhibition that indicates the capabilities of interactions of reactants and generation of products (Buker et al., 2019; Hasanah et al., 2016). It indicates that in an enzyme catalyzed reaction when there is a large excess of substrate and the enzyme concentration is held constant, if substrate concentration is plotted against velocity or reaction rate, a hyperbolic curve is obtained. It reflects that the affinity of the enzyme for its substrate and represents a particular enzyme-substrate system. The smaller the value of Km explained the extra strongly the enzyme binds to the substrate. Kinetic values of enzyme catalyzed reactions are generally considering under the steady-state conditions that described by Vmax and Km (Sjögren et al., 2009; Zaharudin et al., 2019). Consequently, the significant interactions shown by molecular docking through the availabilities of numbers of hydrogen bonds, potential binding energy, and interacting residues of the caffeic acid and pravastatin with target protein (HMGCR). Furthermore, the assessments of MDs revealed the active binding of the compound and protein in a stable manner without any position alteration from the active cavity. The several studies reported and suggested that the protein and ligands interactions depend upon the capabilities of functional groups by the numbers of H-bond, binding energy, and related interactions of small molecule phytocompounds and terminal catalytic residues of target protein (Jasmine and Vanaja, 2013; Makegowda et al., 2019). Along with this, caffeic acid and pravastatin performed optimised ranged parameters of ADMET and the drug likeness by following the five rules of the Lipinski. It well established fact that the BBB effectively regulates the selective drive of cells, ions, fluid, nutrients between the blood and the brain which plays a vital role of protecting the brain or vital organs against potential toxins, encephalitogenic cells, and microbial pathogens, to maintain cerebral homeostasis that is required for proper neuronal and synaptic functions (Jia et al., 2020; Tian et al., 2015). The value of iLogP of the caffeic acid and pravastatin showed significant lipophilicity and hydrophobicity which indicate suitable bioavailability as reported by numerous studies (Daina et al., 2014).
CONCLUSION
Based on the determinations of the study, it can be concluded that caffeic acid has the capability to inhibit the HMGCR activity which may be useful for therapeutics of hypercholesterolemia by subsiding the cholesterol biosynthesis. Further validations can be confirmed from the in-vivo assessments of biochemistry and molecular biology.
ACKNOWLEDGEMENT
The authors sincerely acknowledge the department of pharmacology, AIIMS, Jodhpur, for supporting the in-vitro assay of HMGCR as well as the team of Prof. Surajit Gosh for the in-silico investigations.
CONFLICT OF INTEREST
There was none of conflict of interest.
FUNDING
This work was not supported by any kind special research grant.
AUTHOR CONTRIBUTIONS
HR & SG-designed and review the study. KS & AC—Molecular docking and in-vitro assessments. CK-first rough draft. RSR-MDs.
FUNDING
There is no funding to report.
REFERENCES
Ademosun AO, Oboh G, Passamonti S, Tramer F, Ziberna L, Boligon AA, Athayde ML. Phenolics from grapefruit peels inhibit HMG-CoA reductase and angiotensin-I converting enzyme and show antioxidative properties in endothelial EA.Hy 926 cells. Food Sci Hum Wellness [Internet], 2015; 4:80–5. CrossRef
Agunloye OM, Oboh G. Modulatory effect of caffeic acid on cholinesterases inhibitory properties of donepezil. J Complement Integr Med, 2018; 15:1–10. CrossRef
Anwar S, Shamsi A, Shahbaaz M, Queen A, Khan P, Hasan GM, Islam A, Alajmi MF, Hussain A, Ahmad F, Hassan MI. Rosmarinic acid exhibits anticancer effects via MARK4 inhibition. Sci Rep, 2020; 10:1–14. CrossRef
Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res, 2018; 46:W257–63. CrossRef
Brooks CL, Case DA, Plimpton S, Roux B, Van Der Spoel D, Tajkhorshid E. Classical molecular dynamics. J Chem Phys, 2021; 154:100401–5. CrossRef
Buker SM, Boriack-Sjodin PA, Copeland RA. Enzyme—inhibitor interactions and a simple, rapid method for determining inhibition modality. SLAS Discov, 2019; 24:515–22. CrossRef
Daina A, Michielin O, Zoete V. iLOGP: a dimple, robust, and efficient description of n?octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model, 2014; 54:3284–301. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1–13. CrossRef
Daina A, Zoete V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem, 2016; 11:1117–21. CrossRef
de Alencar Silva A, Pereira-de-Morais L, Rodrigues da Silva RE, de Menezes Dantas D, Brito Milfont CG, Gomes MF, Araújo IM, Kerntopf MR, Alencar de Menezes IR, Barbosa R. Pharmacological screening of the phenolic compound caffeic acid using rat aorta, uterus and ileum smooth muscle. Chem Biol Interact, 2020; 332:1–9. CrossRef
Hasanah Q, Faridah DN, Andrianto D. Inhibition activity of HMG-CoA reductase by rice brain extract and its fractions as anticholesterolemia in vitro study. Der Pharma Chem, 2016; 8:1–5.
Holdgate GA, Meek TD, Grimley RL. Mechanistic enzymology in drug discovery?: a fresh perspective. Drug Discov Rev, 2017; 17:115–32. CrossRef
Iheagwam FN, Ogunlana OO, Ogunlana OE, Isewon I, Oyelade J. Potential anti-cancer flavonoids isolated from Caesalpinia bonduc Young twigs and leaves: molecular docking and in silico studies. Bioinform Biol Insights, 2019; 13:1–16. CrossRef
Mani JS, Johnson JB, Steel JC, Broszczak DA, Neilsen PM, Walsh KB, Naiker M. Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res, 2020; 284:1–16. CrossRef
Jasmine JM, Vanaja R. In silico analysis of phytochemical compounds for optimizing the inhibitors of HMG CoA reductase. J Appl Pharm Sci, 2013; 3:43–7.
Jia CY, Li JY, Hao GF, Yang GF. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today, 2020; 25:248–58. CrossRef
Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov, 2005; 4:206–20. CrossRef
Kumar A. A review of hyperlipidemia and medicinal plants. Int J Appl Pharm Sci Bio-Medical Sci, 2016; 2:219–37.
Laranjinha J, Cadenas E. Redox cycles of caffeic acid, α-tocopherol, and ascorbate: implications for protection of low-density lipoproteins against oxidation. IUBMB Life, 1999; 48:57–65. CrossRef
Liang G, Kou H, Wang T, Guo Y, Ping J, Wang H. Optimization, validation and application of spectrophotometric assay for 3-hydroxy-3-methylglutaryl- coenzyme a reductase activity. Trop J Pharm Res, 2015; 14:671–7. CrossRef
Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des, 2013; 27:221–34. CrossRef
Makegowda M, Doddarevanna RH, Mukundaswamy CK. Molecular docking and multitudinous spectroscopic studies to elucidating proton-pump inhibitor a lansoprazole binding interaction with bovine serum albumin. Biointerface Res Appl Chem, 2019; 9:4015–21. CrossRef
Marahatha R, Basnet S, Bhattarai BR, Budhathoki P, Aryal B, Adhikari B, Lamichhane G, Poudel DK, Parajuli N. Potential natural inhibitors of xanthine oxidase and HMG-CoA reductase in cholesterol regulation: in silico analysis. BMC Complement Med Ther, 2021; 21:1–11. CrossRef
McKenney JM. Pharmacologic characteristics of statins. Clin Cardiol, 2003; 26:32–8. CrossRef
Mukherjee K, Biswas R, Chaudhary SK, Mukherjee PK. Botanicals as medicinal food and their effects against obesity. In Evidence-Based Valid Herb Med, (ed) Pulok K Mukherjee, Elsevier, Kolkata, India, pp 373–402, 2015. CrossRef
Noha SM, Schmidhammer H, Spetea M. Molecular docking, molecular dynamics, and structure-activity relationship explorations of 14-oxygenated n-methylmorphinan-6-ones as potent μ-opioid receptor agonists. ACS Chem Neurosci, 2017; 8:1327–37. CrossRef
Park JB. 5-Caffeoylquinic acid and caffeic acid orally administered suppress P-selectin expression on mouse platelets. J Nutr Biochem, 2009; 20:800–5. CrossRef
Rastogi S, Pandey MM, Rawat AKS. Traditional herbs: a remedy for cardiovascular disorders. Phytomedicine, 2016; 23:1082–9. CrossRef
Rizvi SM, Shakil S, Haneef M. A simple click by click protocol to perform docking?: Autodock 4.2 made easy for non-bioinformaticians. Excli J, 2013; 12:831–57.
Saeedi Saravi SSS, Saeedi Saravi SSS, Arefidoust A, Dehpour AR. The beneficial effects of HMG-CoA reductase inhibitors in the processes of neurodegeneration. Metab Brain Dis, 2017; 32:949–65. CrossRef
Salvamani S, Gunasekaran B, Shukor MY, Shaharuddin NA, Sabullah MK, Ahmad SA. Anti-HMG-CoA reductase, antioxidant, and anti-inflammatory activities of Amaranthus viridis leaf extract as a potential treatment for hypercholesterolemia. Evid Based Complement Altern Med, 2016; 2016:1–10. CrossRef
Sjögren E, Lennernäs H, Andersson TB, Gråsjö J, Bredberg U. The multiple depletion curves method provides accurate estimates of intrinsic clearance (CLint), maximum velocity of the metabolic reaction (Vmax), and Michaelis constant (Km): accuracy and robustness evaluated through experimental data and Monte Carlo si. Drug Metab Dispos, 2009; 37:47–58. CrossRef
Srinivas NR. Dual Incorporation of the in vitro data (IC50) and in vivo (Cmax) data for the prediction of area under the curve (AUC) for statins using regression models developed for either pravastatin or simvastatin. Drug Res (Stuttg), 2016; 66:402–6. CrossRef
Tian S, Wang J, Li Y, Li D, Xu L, Hou T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev, 2015; 86:2–10. CrossRef
Tsai SJ, Chao CY, Yin MC. Preventive and therapeutic effects of caffeic acid against inflammatory injury in striatum of MPTP-treated mice. Eur J Pharmacol, 2011; 670:441–7. CrossRef
Wang Y, Zhang G, Pan J, Gong D. Novel insights into the inhibitory mechanism of kaempferol on xanthine oxidase. J Agric Food Chem, 2015; 63:526–34. CrossRef
Zaharudin N, Staerk D, Dragsted LO. Inhibition of α-glucosidase activity by selected edible seaweeds and fucoxanthin. Food Chem, 2019; 270:481–6. CrossRef
Zeytünlüo?lu A, Zihnio?lu F. Evaluation of some plants for potential dipeptidyl peptidase IV inhibitory effects in vitro. Turkish J Biochem, 2015; 40:217–23. CrossRef