
INTRODUCTION
Vanishing bile duct syndrome (VBDS) is a rare and serious disorder that results from various causes, including neoplasms, infections, congenital and genetic diseases, and drugs (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). Most of these cases are associated with recurrent antibiotic use. The drugs commonly causing VBDS are antibiotics (penicillins, cephalosporins, fluoroquinolones, sulfonamides, macrolides, lincomycins, tetracyclines, and carbapenems), anti-virals (nevirapine), anti-fungals (thiabendazole), anti-helmetic (thiabendazole), anti-malarials (atovaquone-proguanil), and anti-convulsants (carbamazepine, oxycarbazine, phenytoin, valproic acid, phenobarbitone, lamotrigine, and zonisamide) (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). Non-steroidal anti-inflammatory drugs (NSAIDs), sex hormones, antidepressants, and immunomodulators are also responsible for the development of the other class of drugs which can lead to VBD (Oppenheimer et al., 2013).
This condition presents with clinically marked chronic cholestatic liver disease and it can also present with loss of intrahepatic bile ducts. VBDS can also occur following severe cholestatic liver disease, immune–allergic features like edema of the face, fever, rashes, eosinophilia, and lymphadenopathy, and in worse-case scenario with toxic epidermal necrolysis or Stevens–Johnson syndrome (SJS). The exact pathophysiology of VBDS is still unknown. It is believed that it is caused by the immune response triggered by the drug, which is generally dose-independent. As a result of this immune response, invasion and disappearance of the interlobular bile ducts occur which will then lead to chronic cholestasis (Zhao et al., 2017).
The diagnosis of VBDS is mainly based on clinical and pathological presentations (Zhao et al., 2017). Hepatic pathology and liver biopsy are required even in the milder forms of the syndrome. Evidences that can contribute to the confirmation of diagnosis VBDS includes:
- Persistently elevated serum alkaline phosphatase (ALP) and bilirubin for more than 6 months after the initiation of drug-induced liver disease.
- Persistently elevated serum ALP and/or gamma-glutamyl transpeptidase (GGT) for more than 12 months after the initiation of drug-induced liver disease.
- There should be no clinical or serologic evidence suggesting of primary biliary cholangitis, sclerosing cholangitis, and graft-versus-host disease.
- Liver biopsy that shows ductopenia that is taken at least 1 month after the initiation of drug-induced liver disease (National Institute of Diabetes and Digestive and Kidney Diseases, 2012; Oppenheimer et al., 2013).
The clinical presentation of a patient with VBDS includes chronic pruritus, fatigue, and jaundice. In some instances, patients also exhibit very high lipid levels, dyslipidemia, hypercholesterolemia, and skin xanthomata. Skin xanthomata, when present on palms and soles, can be extremely painful and can interfere with daily activities. Mild pruritis can be managed with anti-histamines and in few cases cholestyramine and colestipol, which are bile acid resins, may be useful as they bind and trap pruritogenis in the intestine. Liver transplantation is indicated if severe intractable pruritus is confirmed (National Institute of Diabetes and Digestive and Kidney Diseases, 2012).
Yet, successful therapy that can induce the regeneration of the bile duct that is not available. Current management focuses on improving cholestasis and suppressing the immune reaction (Desmet, 1997). The treatment of VBDS and severe cholestasis includes corticosteroids, but there is lack of evidence regarding its benefits, since they may lead to secondary metabolic effects of cholestasis and end-stage liver disease. Treatment with Ursodiol is universal in VBDS, but their benefit has not been demonstrated through any prospective control trials (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). In few cases, progressive bile duct loss may lead to liver failure, biliary cirrhosis, liver transplantation, or even death (Desmet, 1997). This review focuses on the clinical findings of VBDS and the treatment strategies for the same.
MATERIALS AND METHODS
A detailed review was conducted using the available databases, such as PubMed, PLOS, and ScienceDirect, using search words such as cholestatic disease, VBDS, pediatrics, corticosteroids, and liver transplantation. The articles were thoroughly scrutinized and seven relevant case-based studies were taken and discussed in this review. This review discusses the evidence of VBDS in pediatrics and its management.
Case 1
A female child aged 10 years was diagnosed with rhinopharyngitis and received a pediatric dose of ibuprofen. Eight days later, the patient presented with a relapse of fever and was treated again with ibuprofen. Two days past the first episode of relapsed fever, the child developed maculopapular pruriginous rashes over her face. After 48 hours, she had the appearance of rashes and thus she was admitted in hospital with complaints of dark colored urine, cheilitis, pharyngeal erythema, jaundice, discolored stools, and maculopapular rash with small bullae like features throughout the body. A confirmatory diagnosis of VBDS was made based on laboratory investigations. She was initiated with ursodeoxycholic acid (UDCA) (600 mg/m2/day or 25 mg/kg/day) and liposoluble vitamins. Rifampicin was started later for persistent pruritus. After 10 months, the skin lesions disappeared. Two years later, on follow-up, the child happened to have normal physical status and activity. UDCA therapy was continued (Taghian et al., 2004).
Case 2
A 12-year-old male child with a history of focal seizures on carbamazepine therapy developed a maculopapular rash, which became normal after stopping carbamazepine. Later, sodium valproate was added to the therapy and it did not help the child to control the seizures, and he was prescribed lamotrigine. The child developed a macular rash after 2 weeks of lamotrigine and the rash subsided after the withdrawal of the drug. The rash started subsiding after 1 month of starting lamotrigine and the child developed conjugated hyperbilirubinemia. Laboratory data revealed hyperbilirubinemia. Later, a diagnosis of drug-induced cholestasis was made. He was put on UDCA and vitamin supplements. The child was showing clinical improvements in the beginning and later the condition had worsened as jaundice and pruritus increased. His biochemical markers were increased and ductular proliferation with a paucity of interlobular bile ducts was revealed by liver biopsy and biopsy also showed evidence of bridging fibrosis, and a diagnosis for VBDS was considered. The child was initiated and continued with prednisolone (2 mg/kg) and azathioprine (1.5 mg/kg/day). Given his worsening liver function, he was considered for liver transplant (Bhayana et al., 2012).
Case 3
A 13-year-old female child developed fever and nausea and was treated with diclofen and loxoprofen for 2 and 1 days, respectively. Her provisional diagnosis was jaundice initially and then a confirmatory diagnosis of VDBS due to NSAIDS was confirmed. She was not responding to UDCA and it was decided to carry out plasmapheresis. After plasmapheresis, her serum bilirubin decreased and lymphocyte-stimulating test showed negative for diclofenac and loxoprofen. A liver biopsy was carried out after 6 months and in the portal areas, evidence of regenerated bile ducts was found. This case report showed the evidence of diclofen and loxoprofen-induced VBS for the first time (Zhao et al., 2017).
Case 4
A 10-year-old male child with a history of mild asthma, attention-deficit hyperactivity disorder (ADHD), and depression presented with new complaints of jaundice. He developed bilateral, neck and facial swelling and was treated with prednisone 20 mg and antihistamine 7 days before admission. His medical history included cutaneous larva migrans for which he was given mebendazole 100 mg twice daily for 3 days. He developed dyspnea, rhinitis, fever, cough, and upper respiratory symptoms for which he was initiated with albuterol and amoxicillin-clavulanate 600 mg twice daily for 5 days. He later developed anorexia, icterus, nausea, fatigue, and dark-colored urine. He had a history of dark urine, abdominal pain, and pruritus. His daily medications included Clonidine 0.1 mg for ADHD, Risperidone 0.5 mg, and Citalopram 20 mg. On examination, jaundice and scleral icterus without xanthomas were present. The abdomen examination showed right upper quadrant tenderness and there was no ascites or hepatosplenomegaly.
Provisional diagnosis was made as cholestasis, hepatitis, and pancreatitis with a concurrent upper respiratory tract infection. With supportive treatment, the pancreatits resolved but scleral icterus, jaundice, and serum bilirubin level raised persistently. After 7 days, amoxicillin-clavulanate was discontinued and UDCA 15 mg/kg/day was initiated to the therapy. He had progressive cholestasis with severe pruritus. Canalicular cholestasis with ductopenia was revealed by a liver biopsy after 1 month. Chronic periportal inflammation was apparent with lymphocytes, mild fibrosis, and scattered eosinophils. Naltrexone, lorazepam, diphenhydramine, and hydroxyzine were given for pruritus, and to prevent vitamin deficiencies vitamin supplements (A, D, E, and K) were given. After a month of symptoms, cholestyramine 2 g orally and UDCA dose was raised to 30 mg/kg/day for 6 weeks. For intractable pruritus, the UDCA dose was further raised to 45 mg/kg per day. Adjunct therapy with rifampin 150 mg twice daily was added. Total bilirubin (TB) significantly decreased within 2 weeks after the discontinuation of Cholestyramine. Four months later, scleral icterus, jaundice, and pruritus were completely resolved and the patient became symptom-free after 2 years (Kawasaki et al., 2013).
Case 5
A 7-year-old girl with acute viral infection, after 4 days, presented with fever and was admitted to the hospital. She was suffering from complicated pulmonary edema, myocarditis, and sepsis and was put on ventilatory support. Trimethoprim-sulfamethoxazole and ceftazidime were given for 14 days and doxycycline for 7 days was administered due to leptospirosis and scrub typhus. After 2 weeks, she developed fever, generalized erythematous papules in the oral cavity, genitalia, anus, and conjunctivae. Drug hypersensitivity reaction was diagnosed and dexamethasone was administered. One week after the onset of rash, cholestasis was noted. Icteric sclerae, jaundice in non-desquamated areas, a generalized maculopapular rash with blistering on her neck and in extremities. Intravenous immunoglobulin was administered for 3 days at a dose of 0.5 g/kg along with prednisolone (2 mg/kg/day) and UDCA (20 mg/kg/day). The diagnosis of VBDS was established. There was no clinical or biochemical improvement after starting prednisolone for 15 days and methylprednisolone was then administered for the next 3 days. A repeated liver biopsy was carried out. At 8 weeks, Cyclosporin A 4 mg/kg/day was administered due to failure of intralobular bile ducts. Twenty weeks from the onset of the symptoms, the child developed dyspnea of unknown cause. There are no significant other conditions and the child’s condition progressed to chronic liver failure disease for which liver transplantation was considered (Karnsakul et al., 2006).
Case 6
A 6-year-old patient presented with jaundice and vesiculobullous rash. The patient’s previous medical history showed evidence of drug-induced rash due to the administration of naproxen and amoxicillin. On physical examination, the patient showed severe hepatomegaly, jaundice, and a year old vesiculobullous rash, throughout his body. Diagnosis of SJS-induced amoxicillin and naproxen was made. The patient was treated with methylprednisolone for 2 mg/kg per day for 15 days, plasma exchange (total of five exchanges), and supportive care at the first hospital. The patient’s dermatological lesions improved, but the cholestasis continued even after 16 days of treatment.
In the second hospital, IV methylprednisolone, followed by oral methylprednisolone, was continued for a total of 6 weeks (2 mg/kg per day) along with UDCA (15 mg/kg per day, orally) and traditional Chinese supportive medicine care. Cholestasis marker levels had steadily increased after 6 weeks of treatment. A liver biopsy was carried out and the biopsy specimen revealed lobular structures and the interlobular bile duct was visible only in three portal areas. Interlobular bile duct epithelium showed marked degeneration. CK7 staining was positive; there were lobular capillary bile plugs; liver cell necrosis was observed. An assessment of VBDS was made. UDCA 40 mg/kg per day, orally, S-adenosyl-L-methionine 1,000 mg per day, IV, alprostadil 10 μg per day, IV, and Pien Tze Huang traditional Chinese medicine were administered. The patient was discharged and followed-up at the clinic for treatment with UDCA and Pien Tze Huang. After 5 months, the patient’s condition improved and the liver function was normalized. On follow-up, the patient’s condition improved and his liver function returned to normal (Li et al., 2019).
Case 7
A 7-year-old boy was hospitalized with complaints of jaundice. His past medical history showed he had diarrhea and mild fever 2 weeks before and he was prescribed Trimethoprim 80 mg and Sulfamethoxazole 400 mg. After 8 days, he showed symptoms of jaundice and severe pruritus. Three months prior to the diagnosis of jaundice, he received antibiotic therapy with amoxicillin-clavulanic acid for 5 days for acute tonsillitis in a private clinic. Treatment with UDCA 20 mg/kg/day was started and continued. Thirty-six days after discharge, liver biopsy was carried out as the patient was not showing any improvement of cholestasis. Histologic examination revealed disappearance of the interlobular bile ducts along with bile stasis and biliary plugs around the central vein. He was diagnosed with VBDS and the dose of UDCA was increased to 30 mg/kg/day. He was given with amoxicillin-clavulanic acid for a week due to acute rhinosinusitis which he developed 3 months after the recovery of VBDS. The patient has been followed-up continuously as an outpatient at the clinic (Cho et al., 2013).
RESULTS AND DISCUSSION
VBDS is a severe form of the cholestatic disease which is associated with certain medications like, lamotrigine, carbamazepine, NSAIDs like acetaminophen, naproxen, ibuprofen, sertraline, and antibiotics. More than a hundred different medications play a role in developing VBDS. This condition usually begins within 1–14 days after the exposure to causative agent but may not manifest for up to 3–6 weeks after ingestion. In the pediatric population, VBDS is a rare condition and reported cases are sparse. The adult population is more likely to have VBDS due to multiple causes of liver injury. In all the above-mentioned case reports, the pediatric population was affected with SJS. SJS is a well-known immune complex-mediated hypersensitivity reaction that affects all, irrespective of age. In adults, due to complications of acute drug-induced liver injury, VBDS generally manifests within 1–6 months after liver injury. Of all hospitalizations due to jaundice, 2–5% is due to drug toxicity. The classical confirmatory test is identified to be a liver biopsy. Absence or diminished intra-hepatolobular ducts at least in 10 portal sites in biopsy samples indicates ductopenia. Liver ultrasonography and abdominal CT scans are the two unique diagnostic tests that provide a visual image of the hepatic enhancement pattern in patients with VBDS. Other significant markers are the presence of elevated liver function tests such as aspartate transaminase (AST) or alanine transaminase (ALT), TB or direct bilirubin, ALP, GGT, albumin, and total cholesterol level. The mechanism of interlobular duct loss and biliary epithelial cell injury in the VBDS has not been fully established (Goldman’s Cecil Medicine, 2012; Srivastava et al., 1998; Taghian et al., 2004; Zhao et al., 2017).
Pathogenesis
Few studies propose metabolic, toxic, immune, and idiosyncratic etiologies as possible factors which could aggravate VBDS. Based on available data, a hypothesis of the pathophysiology has been generated and shown in Figure 1. More studies must be conducted at the histopathological level to sketch the exact mechanism of how SJS could lead to the development of VBDS (Goldman’s Cecil Medicine, 2012).
The bile duct disappears because of pathologic manifestation caused by multiple diseases and mechanisms. In healthy liver, the biliary epithelial layer is maintained through the balance between cell degeneration and regeneration, unlike other body tissues. Cell death occurs via a well-studied mechanism known as apoptosis. The biliary tree expresses the proteins responsible for the regulation of apoptosis (Oppenheimer et al., 2013). Apoptosis promoters like B-cell lymphoma/leukemia 2 (BCL2)-associated X PROTEIN (Bax) and apoptosis antagonist like BCL2 are expressed in the biliary tree and bile ductules, respectively. Biliary epithelial apoptosis can occur as the result of activation of cell death receptors by various insults (Reau and Jensen, 2008).
Cell-death receptors like CD95 (Fas) and its ligand (FasL), necrosis factor-alpha, perforin and granzyme B, tumor, and oxidative stress can cause damage to the DNA and BCL2 downregulation, which leads to bile duct injury. The presence of allo or autoantigens on biliary epithelial cells produced by immunologic cells initiates a cascade that ultimately results in biliary injury. The CD3-positive T-cells predominate in general, but in some disease states of the dominance of T cells (CD4þ or CD8þ) vary. The presence of an appropriate co-stimulatory signal stimulates the release of proinflammatory cytokines, which can further lead to multiplication of cytotoxic T lymphocytes, more efficient antigen presentation, and immune cell recruitment. Eventually, apoptosis and T-cell cytotoxicity happen, resulting in bile duct loss (Goldman’s Cecil Medicine, 2012; Nakanuma et al., 2001).
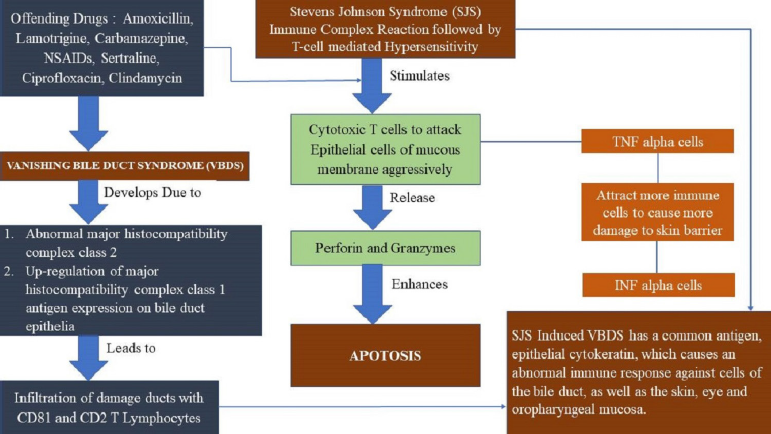 | Figure 1. Proposed hypothesis (13). [Click here to view] |
According to the shreds of evidence, it has been understood that all the pediatric patients developed VBDS as an after-effect of SJS due to certain drugs. In all the cases, the patients were hospitalized after the appearance of jaundice symptoms, maculopapular rashes extending to the whole body, dark urine, discolored stools, arthralgia, conjunctivitis, chapped lips, pruritus, and vesiculobullous rash (Cho et al., 2013; Goldman’s Cecil Medicine, 2012; Li et al., 2019; Taghian et al., 2004). In most of the cases, the children had a previous history of lower respiratory infection for which they were administered antibiotics and NSAIDs. On laboratory findings, all the cases were found to have an elevated liver test, such as ALT, AST, ALP, GGT, bilirubin (total and direct), and serum cholesterol. Coagulation function and complete blood test results were normal in all patients. Electrolyte levels and renal function tests were within normal limits. In few patients, CT scans showed the appearance of hepatomegaly.
How to treat VBDS effectively?
Due to the lack of established treatment guidelines for VBDS, the patients were treated with UDCA, methylprednisolone, rifampin, plasmapheresis, S-adenosyl-L-methionine, azathioprine, and cholestyramine, along with removing the offending agent and providing supportive care (Bhayana et al., 2012).
Prompt recognition and removal of the offending agent
It is necessary to review the patient’s medication to evaluate and ensure that the reaction has occurred due to any medication that the patient has taken. A severe cholestatic disease like VBDS is often fatal or life-threatening. Hence, the drug must be discontinued and should not be rechallenged (Schatz and Weber, 2015).
Initiation of supportive care
The primary goal of supportive care is to prevent and treat the symptoms of a disease or adverse reaction to certain drugs. This would improve the quality of life from further deterioration of the life-threatening condition (National Cancer Institute, 2020).
First-line therapeutic options
Ursodeoxycholic acid (UDCA)
There are studies that say UDCA acts by various mechanisms, including protection of the hepatocellular lipid membranes against hydrophobic cytotoxic bile acids, choleresis, and downregulation of bile acid-induced apoptosis of the liver, therefore exerting therapeutic action. UDCA is a hydrophilic dihydroxy bile acid compound that helps in the acceleration of transcription by the transporter protein (Goldman’s Cecil Medicine, 2012). UDCA use in the pediatric population for 6–7 months led to a drastic improvement in liver function. All the bile parameters were normalized in children.
The conventional pediatric dose in treating pediatrics ranges from 20 to 30 mg/kg/day (Bhayana et al., 2012; Cho et al., 2013; Taghian et al., 2004). UDCA restores bile blow, improves the canalicular transport, promotes the expulsion of toxic bile salts from the hepatic cells, and prevents hepatocytes from apoptosis and necrosis. UDCA decreases biliary expression of MHC class II molecules that might be suggestive for reducing T-cell-medicated hepatocellular damage (Desmet, 1997).
Co-administration of ursodeoxycholic and cholestyramine leads to reduced bioavailability of UDCA and results in intraluminal binding. Thus, leads to diminished intestinal uptake of UDCA (Kawasaki et al., 2013). Therefore, the use of cholestyramine could be avoided.
Rifampin
Rifampin improves and relieves pruritus. The conventional pediatric dose in treating pediatrics can range from 4 to 10 mg/kg/day. It helps in the treatment of pruritis by inhibiting the bile acid uptake by the hepatocytes. It can enhance the activity of a mixed function system and increases the level of the hepatic cytochrome P-450. These enhance 6-α hydroxylation and subsequent 6-α glucuronidation of bile acids, facilitating the synthesis of protective acids and decreasing toxic bile acids. The antimicrobial properties of rifampin modify the synthesis of secondary bile acids in the intestinal lumen and consequently reduce the amount of hepatotoxic lithocholic bile acids. Hence, it proves to be beneficial in VBDS (White and Appleman, 2014).
Second-line treatment
Corticosteroids
The treatment of VBDS and severe cholestasis includes corticosteroids, but there is a lack of evidence available regarding its benefit, and they may lead to secondary metabolic effects of cholestasis and end-stage liver disease (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). Hence, corticosteroids must be used with utmost caution.
Plasmapheresis
Some studies suggest the use of plasmapheresis in treating VBDS, but the effectiveness of plasma exchange is to yet to be established.
Immunosuppressant
The use of immunosuppressants in one of the pediatric studies was found to be ineffective and has led to worsening liver functions, including hard liver and coagulogram, on clinical examination which later ended up in liver transplant (Goldman’s Cecil Medicine, 2012).
CONCLUSION
After a thorough literature review, VBDS, especially in the pediatric population, remained unidentified, unless the patients presented with life-threatening symptoms. Concurrent occurrences of SJS and VBDS are very rare. And for this reason, we strongly believe it is necessary to develop an established guideline for treating VBDS in pediatrics, as well as adults. Curative therapies, such as transplants or surgical resection, are not an option for patients who are not in an advanced state of VBDS. Even though UDCA is not identified as the first-line treatment for VBDS in pediatrics, many works in the literature support the use of the drug. UDCA is well tolerated and shows a better response when compared to other medications used conventionally in treating VBDS. In conclusion, we recommend UDCA as the treatment primary treatment once the biopsy shows ductopenia which is the major indicator of VBDS. Further studies should be conducted on the use of immunosuppressants and plasma exchange to provide a better strategy for the treatment of VBDS.
ACKNOWLEDGMENTS
The authors would like to acknowledge the Department of Pharmacy Practice, JSS College of Pharmacy, Ooty, for their support.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICT OF INTEREST
The authors of this paper have no conflicts of interest to declare.
FUNDING
There are no funding organizations that supported this review.
LIST OF ABBREVIATIONS
VBDS Vanishing bile duct syndrome
NSAIDs Non-steroidal anti- inflammatory drugs
UDCA Ursodeoxycholic acid
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Bhayana H, Appasani S, Thapa BR, Das A, Singh K. Lamotrigine-induced vanishing bile duct syndrome in a child. J Pediatr Gastroenterol Nutr, 2012; 55(6):e147–8. CrossRef
Cho HJ, Jwa HJ, Kim KS, Gang DY, Kim JY. Urosodeoxycholic acid therapy in a child with trimethoprim-sulfamethoxazole-induced vanishing bile duct syndrome. Pediatr Gastroenterol Hepatol Nutr, 2013; 16(4):273–8. CrossRef
Desmet VJ. Vanishing bile duct syndrome in drug-induced liver disease. J Hepatol, 1997; 26(1):31–5. CrossRef
Goldman’s Cecil Medicine. Vanishing bile duct syndrome, 24th edition. Library of Congress Cataloging - in Publication Data, Newyork, 2012.
Karnsakul W, Arkachaisri T, Atisook K, Wisuthsarewong W, Sattawatthamrong Y, Aanpreung P. Vanishing bile duct syndrome in a child with toxic epidermal necrolysis: an interplay of unbalanced immune regulatory mechanisms. Ann Hepatol, 2006; 5(2):116–9. CrossRef
Kawasaki Y, Matsubara K, Hashimoto K, Tanigawa K, Kage M, Iwata A, Nigami H, Fukaya T. Nonsteroidal anti-inflammatory drug-induced vanishing bile duct syndrome treated with plasmapheresis. J Pediatr Gastroenterol Nutr, 2013; 57(5):e30–1. CrossRef
Li L, Zheng S, Chen Y. Stevens-Johnson syndrome and acute vanishing bile duct syndrome after the use of amoxicillin and naproxen in a child. J Int Med Res, 2019; 47(9):4537–43. CrossRef
Nakanuma Y, Tsuneyama K, Harada K. Pathology and pathogenesis of intrahepatic bile duct loss. J Hepatobiliary Pancreat Surg, 2001; 8(4):303–15. CrossRef
National Cancer Institute. NCI Dictionary of Cancer Terms. National Cancer Institute, Bethesda, MD, 2020. Available via https://www.cancer.gov/publications/dictionaries/cancer-terms/def/supportive-care
National Institute of Diabetes and Digestive and Kidney Diseases. LiverTox: clinical and research information on drug-induced liver injury. National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2012.
Oppenheimer AP, Koh C, McLaughlin M, Williamson JC, Norton TD, Laudadio J, Heller T, Kleiner DE, High KP, Morse CG. Vanishing bile duct syndrome in human immunodeficiency virus infected adults: a report of two cases. World J Gastroenterol. 2013; 19(1):115–21. CrossRef
Reau NS, Jensen DM. Vanishing bile duct syndrome. Clin Liver Dis, 2008; 12(1):203–17. CrossRef
Schatz SN, Weber RJ. Adverse Drug Reactions. CNS/Pharm Pract, 2015; 25:5–26.
Srivastava M, Perez-Atayde A, Jonas MM. Drug-associated acute-onset vanishing bile duct and Stevens-Johnson syndromes in a child. Gastroenterology, 1998; 115(3):743–6. CrossRef
Taghian M, Tran TA, Bresson-Hadni S, Menget A, Felix S, Jacquemin E. Acute vanishing bile duct syndrome after ibuprofen therapy in a child. J Pediatr, 2004; 145(2):273–6. CrossRef
White JC. Appleman S. Infliximab/Plasmapheresis in vanishing bile duct syndrome secondary to toxic epidermal necrolysis. Pediatrics, 2014; 134(4):e1194–8. CrossRef
Zhao Z, Bao L, Yu X, Zhu C, Xu J, Wang Y, Yin M, Li Y, Li W. Acute vanishing bile duct syndrome after therapy with cephalosporin, metronidazole, and clotrimazole: a case report. Medicine, 2017; 96(36):e8009. CrossRef