
INTRODUCTION
Cancer is a disease caused by abnormal cells in the body's tissues that grow and develop rapidly and become uncontrolled. This disease is known as one of the world’s major public health problems causing morbidity and mortality, and the number of cancer cases reaches more than 19 million (Ferlay et al., 2020). In 2012, the most diagnosed cancer in women was breast cancer (43.3%) and cervical cancer (14%) (Tao et al., 2015). Compared to 2012, the number of cases increased by 24% in 2018. This number is expected to continue to rise (Bray et al., 2018). The high prevalence and increased incidence made urgent research for effective therapies for cancer (Ghoncheh et al., 2016; Parvizpour et al., 2018).
Currently, treatment of some types of breast cancer was conducted using monoclonal antibodies against HER2 (Atapour et al., 2020). The limitation of passive therapy such as high cost, treatment duration, and the possibility of emerging immunological tolerance has not been resolved (Pallerla et al., 2021). As an alternative to monoclonal antibody therapy with a limited clinical benefit over time, vaccines stimulating immune response toward tumor cells are one of the therapeutic strategies as a potential approach for cancer therapy. Active immunotherapy promoted by the tumor-associated antigen vaccine or tumor-specific antigen will increase preexisting resistance to antigens and prolonged activation of the immune system (Benedetti et al., 2017).
Promising vaccine development methods in the genomic era are by using the reverse vaccinology method using bioinformatics and immunoinformatics approach. This reverse vaccinology approach predicts epitopes, an antigen determinant that has an important role in immunity, and its interaction toward human leukocyte antigen (HLA). Through this approach, vaccine development can be carried out based on the target sequence and genomic data without culturing the target organism (Kanampalliwar et al., 2013). Also, this approach was able to accelerate the initial study process of vaccine development and reduce research costs. The predicted protein from this in silico study can then be synthesized for testing on test animals (Khan et al., 2006; Serruto and Rappuoli, 2006).
Cancer prevalence, incidence, and mortality between diverse ethnic populations are very different. The causes for these differences are complex, including intrinsic factors (genetic variation) and extrinsic factors (social, economic, and geographical) (Khan et al., 2017). Understanding cancer risk and the underlying cause is essential in developing research and health care practices that could work for various ethnicities and populations. Therefore, in the vaccine development process, it is important to pay attention to vaccine coverage so that vaccines can cover various ethnicities.
Some proteins are overexpressed in cancer patients and associated with enhanced tumor growth. These overexpressed proteins are the subject of active immunotherapy in vaccination. In this study, we use protein Receptor tyrosine-protein kinase erbB-2 (ERBB2), Mucin-4 (MUC4), and Phosphatase And Tensin Homolog (PTEN) for vaccine development. ERBB2 is a gene encoding growth factor receptor that plays a significant role in breast cancer (Ludovini et al., 2008). ERBB2 is often overexpressed in breast cancer and has been considered as a potential target in cancer therapeutics (Chentoufi et al., 2009). MUC4 is membrane-bound mucin expressed in several epithelial malignancies. Mucin is normally expressed by epithelial cells as a barrier and contributes to lubrication. MUC4 is mucin that is apparently specific to tumor tissue. Recent studies showed that MUC4 differentially expressed in breast cancer cells appears to correlate with prognosis (Hattrup and Gendler, 2008). PTEN protein is involved in the regulation of several crucial cell functions. Partial loss of PTEN function is enough to promote tumorigenesis and accelerate cancer progression (Bazzichetto et al., 2019). Mutations in these proteins lead to the variability in sequence composition. Conserved sequence regions are critical to addressing the diversity of the proteins (Khan et al., 2008).
This study will be carried out on multiepitope characterization of potential T cells against protein mechanisms in breast cancer, diversity analysis of protein sequences related to immunity, effects of antigenic variation, and sequence similarity to the immune response, HLA. The urgency of this study is based on the high incidence of breast cancer in Indonesia and breast cancer drug resistance. Another alternative is needed to prevent and treat breast cancer by utilization of vaccines.
METHODS
The methodology used in this study is summarized in a diagram (Fig. 1).
Data retrieval
ERBB2 (P04626), PTEN (P60484), and MUC4 (Q99102) protein sequences were retrieved from the UniProt web server (https://www.uniprot.org/) (Bateman, 2019). Mutation data and ERBB2, PTEN, and MUC4 protein sequences were downloaded from the COSMIC website (https://cancer.sanger.ac.uk/cosmic) (Tate et al., 2019). The mutation data downloaded are the missense type mutation data. The mutations were induced in the retrieved sequence by BioEdit (Hall, 1999).
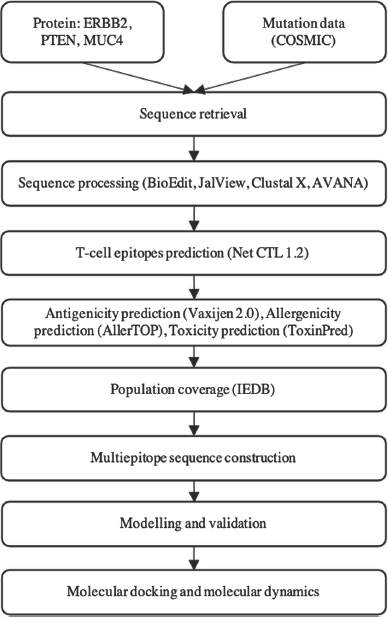 | Figure 1. The methodology of the research. [Click here to view] |
Sequence processing and diversity analysis
After obtaining a sequence with a mutation according to COSMIC data, the sequence is then processed through the JalView software to delete the twin sequences so that the sequences obtained are unique (Waterhouse et al., 2009). The sequences were then aligned using the ClustalX software (Larkin et al., 2007). Mutation in this protein appears to be associated with developing breast cancer. As the gene has a high mutation number based on the database, it is important to search regions of protein which are conserved. The sequences were then analyzed for entropy using the AVANA software to determine the conserved region.
Prediction of the epitope and its characteristics
T-cell epitope prediction was carried out on the NetCTL 1.2 page (https://services.healthtech.dtu.dk/service.php?NetCTL-1.2) based on 12 HLA class I supertypes (A1, A2, A3, A24, B7, B8, B27, B44, B58, B62, C1, and C4) (Larsen et al., 2007). The prediction results with NetCTL 1.2 will provide data on the affinity value of HLA class I and C terminal binding, the value of transport effectiveness via TAP, and the total affinity value (Grifoni et al., 2020). The epitope chosen is an epitope with a value above 0.75. Epitopes obtained from the NetCTL page predicted their immunogenicity characteristics through the IEDB Immunogenicity page (http://tools.iedb.org/immunogenicity/) (Calis et al., 2013).
Immunogenicity and antigenicity are important parameters in vaccine design. Therefore, epitopes that have been obtained from the predictions via the NetCTL 1.2 were submitted to the IEDB immunogenicity page. Epitopes with positive immunogenicity values ​​were further predicted for their antigenicity characteristics through the VaxiJen 2.0 page (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova and Flower, 2007), allergenic characteristics on the AllerTOP page (https://www.ddg-pharmfac.net/AllerTOP/) (Dimitrov et al., 2013), and the characteristics of the toxicity on the ToxinPred page (http://www.imtech.res.in/raghava/toxinpred/) (Gupta et al., 2013). VaxiJen and AllerTOP, respectively, identify the antigenicity and allergenicity of epitopes based on physicochemical properties of proteins (Dimitrov et al., 2013; Doytchinova and Flower, 2007). In the antigenicity prediction test using VaxiJen 2.0, 0.5 was kept as a threshold score and epitopes with a higher score were considered for further analysis. Toxicity prediction by ToxinPred was based on support vector machine from toxic peptides dataset (Joshi et al., 2020). ToxinPred prediction consists of two steps, toxicity prediction and analog generation and prediction (Gupta et al., 2013). Epitopes selected as candidates are epitopes with an antigenicity value of more than 0.5 and nontoxic and nonallergen characteristics.
Epitope population coverage prediction
The coverage of the CTL epitope population is predicted via the IEDB Population Coverage page with default parameters, based on Major Histocompatibility Complex (MHC) binding restriction data (http://tools.iedb.org/population/). This dataset consists of frequencies for 115 countries, 21 ethnicities, and 16 geographical areas. Each CTL epitope is inserted to that page along with the HLA type data that binds to the epitope. After that, select the area and population to predicts epitope population coverage. The areas selected are world, East Asia, Northeast Asia, South Asia, Southeast Asia, Southwest Asia, Europe, Central Africa, East Africa, North Africa, South Africa, West Africa, West Indies, Central America, North America, South America, and Oceania (Bui et al., 2006).
Multiepitope sequence construction
Each of the selected epitopes was then linked with AAY linker from the study by Klein et al. (2014) and a HEYGAEALERAG motif was added to enhance epitope presentation (Tani et al., 2000).
Epitope modeling
3D modeling of the putative multiepitope vaccine was conducted through Swiss-PdbViewer/DeepView (Guex and Peitsch, 1997). The residues of the structure can be seen through the Control Panel menu. The attachment of the vaccine structure to the template is done through the Magic Fit menu. The adhered residues can be seen through the alignment menu. Visualization of the tertiary structure of the adhered vaccine is done by removing the tertiary structure of the template so that the tertiary structure of the vaccine can be seen. The improvement of the tertiary structure of the vaccine is carried out in two stages. The first step is to improve the structure of the vaccine in the DeepView viewer by fixing the overlapping residues, whereas the second stage is through the Swiss-Model server by sending the structure of each vaccine that has been upgraded on DeepView to the Swiss-Model server in the form of PDB for structure optimization, namely, by selecting the Optimize Mode menu on the Swiss-Model server.
Validation of tertiary structure
The evaluation of tertiary structure of putative multiepitope vaccine was done using Ramachandran plot in DeepView program. Some parameters were used to evaluate the quality of 3D model of putative multiepitope such as MolProbability score, Ramachandran favored, Ramachandran outliner, rotamer outliner value, clash score, C beta deviation, bad bonds, and bad angels (Johansson et al., 2012).
Molecular docking
The initial stage of the molecular docking simulation is 3D modeling of the selected multiepitope sequence. 3D modeling is carried out on the SWISS-MODEL page (https://swissmodel.expasy.org) and assessment for the 3D structure using Ramachandran plot (Gopalakrishnan et al., 2007; Waterhouse et al., 2018). The macromolecule TLR4 was downloaded from the Protein Data Bank (https://www.rcsb.org/) with PDB ID 4R7D (Loyau et al., 2015). Molecular docking was conducted using protein-protein docking in Rosetta web server (https://rosie.graylab.jhu.edu/docking2) (Chaudhury et al., 2011; Lyskov and Gray, 2008; Lyskov et al., 2013). PyMOL software was used for visualization and amino acid interaction evaluation for molecular docking results (Delano, 2002).
Molecular dynamic simulation
The molecular dynamic simulation was executed on the LARMD website (http://chemyang.ccnu.edu.cn/ccb/server/LARMD) (Yang et al., 2020). LARMD is developed based on Amber16 software (Case et al., 2016). Molecular dynamic simulation has workflow as follows: preparation complex protein-ligand, force field generation, minimization, equilibration, production, and result analysis root-mean-square deviation [RMSD], root-mean-square fluctuation [RMSF], B-factor, fraction of native contact, and radius of gyration [Rg] (Best et al., 2013; Chovancova et al., 2012; Grant et al., 2006; McGibbon et al., 2015).
RESULTS AND DISCUSSION
Sequence processing
Bioinformatics and immunoinformatics have been supporting large amounts of biological data in vaccine research. Several tools in this approach have been developed to process sequence and analyze immunological data (Raman et al., 2014). JalView is used to remove identical sequences and generate unique sequences. These unique sequences are aligned using ClustalX. Alignment files are converted into * .taln files by BioEdit. The processed sequence then being analyzed its entropy using AVANA, a software based on the Shannon Entropy concept. Shannon sequence entropy analysis measures the conservation level and variability of the sequence of certain lengths. Conserved peptides with lower entropy scores are more conserved and less random than peptides with higher entropy (Li et al., 2019). The conserved 9-mer peptides from AVANA are then analyzed further for the epitope prediction (Khan et al., 2006).
Prediction of T-cell epitopes
The conserved peptides are used for T cell putative epitopes identification using an immunoinformatics approach. Putative epitopes chosen for further analysis should be antigenic and immunogenic. Antigenicity is the ability of a peptide to bind and interact specifically with the functional binding site of an antibody. Immunogenicity refers to the ability of the peptide to induce an immune response (Choudhuri, 2014).
One of the important stages in peptide-based vaccine development is the prediction of antigenic epitopes that potentially bind to different HLA alleles. Recognition of putative epitopes by T-cell receptor (TCR) will induce an immune response. Putative epitopes that are recognized by TCR will be presented on the surface of Antigen Presenting Cells bound to MHC. There are two types of MHC: MHC Class I (HLA class I) and MHC Class II (HLA class II). MHC or HLA molecules are polymorphic and thousands of HLA are identified. Therefore, in putative epitope vaccine design, selection of putative epitopes that bind to a larger number of HLA supertypes could result in increased population coverage (Patronov and Doytchinova, 2013).
The prediction of T-cell putative epitopes was conducted by data-driven using NetCTL-1.2 tools for CTL (HLA I) putative epitopes. The NetCTL-1.2 tools integrate prediction of peptide MHC Class I binding, proteasomal C terminal cleavage, and TAP transport efficiency for 12 HLA supertypes (Larsen et al., 2007). The peptide that has a proteasomal recognition site is not favorable as a putative epitope because it will undergo degradation during processing. TAP binding and transport efficiency are important because the peptides must be transported by TAP to be presented on MHC Class I. The higher the score, the higher the possibility for the putative epitopes to be transported by TAP (Bhasin, 2004).
Epitopes are obtained from the NetCTL server and then its antigenicity is evaluated using VaxiJen and immunogenicity using IEDB. Antigenicity and immunogenicity are important parameters for vaccine development to ensure that the peptide sequence could induce an immune response (Kalita et al., 2020). Besides that, the peptide sequence has to be nontoxic and nonallergenic (Dimitrov et al., 2013).
The nonantigenic putative epitopes were removed, subjected to predict its toxicity and allergenicity. Toxic and allergenic putative epitopes were removed. The results of the CTL epitope analysis, antigenicity, and immunogenicity of the three proteins are summarized in Table 1. The analysis was also conducted for an epitope-based vaccine which has been evaluated through the clinical phase, such as E75 and GP2 vaccine with amino acid composition, respectively, KIFGSLAFL and IISAVVGIL. Both of these vaccines induce CD8+ CTL response (Brown et al., 2020).
Despite the fact that extensive research has been done in epitope-based peptide-vaccine development, the vaccine has not yet been approved for human use. This is due to the low immunogenicity of the simple peptide vaccine and its HLA-restricted characteristics. Hence, it is necessary to construct a multiepitope vaccine targeting different HLAs. In addition, the utilization of adjuvants or certain motifs is used to enhance the immunogenicity of the vaccine (Purcell et al., 2007; TopuzoÄŸullari et al., 2020). Based on the results of the CTL epitope prediction in Table 1, a multiepitope sequence is constructed as a vaccine candidate, with an arrangement as shown in Figure 2. Each epitope is connected to an AAY linker and added to a motif HEYGAEALERAG at the N end to enhance epitope presentation (Safavi et al., 2019). The motif and epitopes were linked by the EAAAK linker.
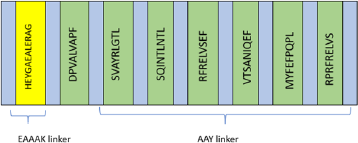 | Figure 2. Multiepitope vaccine constructs. [Click here to view] |
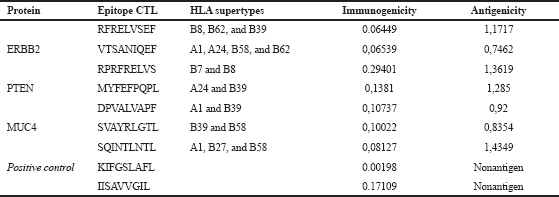 | Table 1. Putative epitope prediction results. [Click here to view] |
Epitope population coverage prediction
MHC or HLA molecules are polymorphic and more than thousands of HLA are identified. In humans, more than 225 HLA Class I and 980 HLA Class II alleles have been identified. The regional polymorphisms encoding the peptide-binding pathways of the HLA molecule give MHC molecules varying binding specificities. In addition, the frequency of allele variants varies among different ethnicities. It would be complex if different epitopes were produced to cover different ethnicities or regions (Sakib et al., 2014). Therefore, it is important to select a putative epitope with a broad and maximal population coverage in addition to the ability to bind HLA.
IEDB population coverage tools generate a prediction of population coverage from the vaccine construct for all regions available and the world, as presented in Figure 3. From the graphic, the vaccine constructs showed good percentage coverage for all regions/populations available in IEDB webserver. Maximum coverage (83.72%) was found in the population of Oceania. Population coverage from the world population was 76.59%. All of the percentage coverage was above 55%; only one population, Central America population, has 2.18% population coverage. The small population coverage was in the Central America region because the Central America region provided scarce information from very specific populations (Michel-Todó et al., 2019).
3D molecular prediction of putative multiepitope vaccine
The homology approach was used in research for the tertiary structure predictions of vaccines. The tertiary structure prediction of a protein with homology modeling can be done in terms of the similarity of the sequence between the protein and the database protein which is at least 20% (Bourne, 2003). The results of the homology modeling sequence of the multiepitope vaccine with the protein in the database show that it is greater than 50%, so the prediction of the vaccine structure can be done using the homology modeling method.
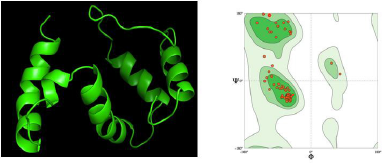
Visualization of the tertiary structure of the reverse immunization vaccine is shown in Figure 4 A. The vaccine design has similar sequences to the TLR4 proteins, which are expected to have the same function and trigger an immune response as in TLR4. Improvements to the tertiary structure of vaccines need to be done to repair residues that overlap each other. Proteins that contain the same amino acid structure as the template do not necessarily fold (folding) proteins in their tertiary form because of the influence of interactions between amino acids with side amino acids or neighboring amino acids. The 3D structure validation is carried out with the Ramachandran plot as shown in Figure 4 B.
The glycine plot can be in the disallowed region because glycine does not have side chains, so the angle φ (phi) and angle ψ (psi) are infinite. The number of residual plots other than glycine in the restricted area indicates the structural quality of the protein. If the amount exceeds 15% of the total protein residue, the protein quality is poor (Cherkasov, 2005). The amount of nonglycine residue in the disallowed region on the Ramachandran plot for the vaccine structure predicted by First Approach Mode and Optimize Mode 0.0% with a clash score of 1.47 is much smaller than 15% of the total residue, which is around 2%–2.5%. Thus, the multiepitope structure of the vaccine produced is of good quality.
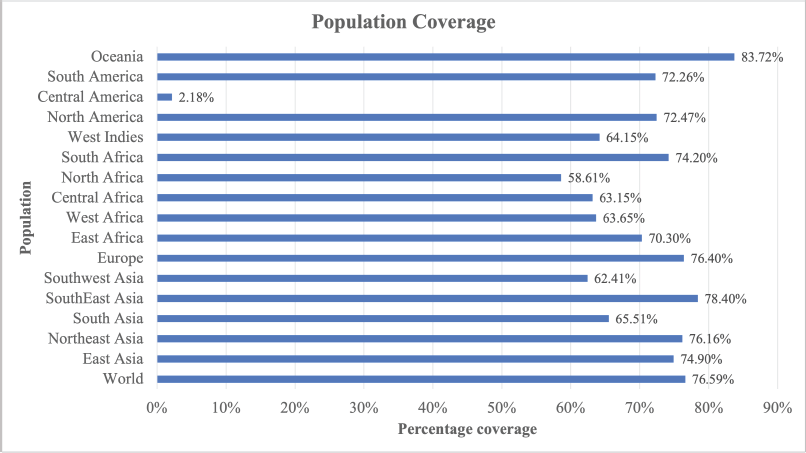 | Figure 3. Population coverage of multiepitope vaccine. [Click here to view] |
The evaluation of the tertiary structure of the vaccine was conducted by looking at the overlapped residues and Ramachandran plot analysis of the vaccine structure integrated with the DeepView (Fig. 4). Certain combinations of φ (phi) and ψ (psi) are not allowed because they will produce a steric hindrance or an overlap between the atoms in the protein. Ramachandran plots are used to check and adjust the conformational measurements of a protein model. The Ramachandran plot has an area marked by a blue line around it, which is the coordinate area for the secondary structure of the protein. This area is the allowed region, where the number of amino acids is plotted in two regions, namely, the yellow area, namely, the area sterically permitting the values of φ (phi) and ψ (psi), and the blue area, which is the area of maximum tolerance. steric strain. Meanwhile, the disallowed region is an area outside the allowed region, where residual plots other than glycine are not allowed in the area.
Based on Ramachandran plot analysis for 3D modeling of putative multiepitope (Fig. 4 A), MolProbability score value is 1.11; Ramachandran favored value is 96.55%; Ramachandran outliner and rotamer outliner value is 0.00%; clash score value is 1.47; C beta deviation is 1 (A75 Asn); bad bonds are 0/705; and bad angels are 5/959 (Arg83, Pro84, Thr51, Asn50, Glu11, Leu43, and Ala44) (Fig. 4 B).
Molecular docking TLR4 with putative multiepitope vaccine
Molecular docking is one of the fundamental structure-based techniques in drug design and bioinformatics, which predicts the interaction between ligand and protein or protein and protein. In immunoinformatics, docking becomes a proper tool to solve the problem of binding prediction for proteins that play a role in the immune system.
Molecular docking analysis using Rosetta web server is a multiscale Monte Carlo-based algorithm that utilized a centroid-based, coarse grain stage to identify favorable docking poses, and an all-atom refinement stage that optimizes rigid-body position and side-chain conformation. RosettaDock has been modified to overcome the critical challenge in protein-protein docking: binding-induced backbone conformational changes. RosettaDock has been applied for antibody-antigen docking research (Sivasubramanian et al., 2006, 2007), peptide docking and specificity (Chaudhury and Gray, 2009; Raveh et al., 2010) multi-body (Bocik et al., 2011) and symmetric-docking (Andre et al., 2008).
 | Figure 4. (A) 3D prediction of putative multiepitope vaccine; (B) Ramachandran plot of putative multiepitope vaccine. [Click here to view] |
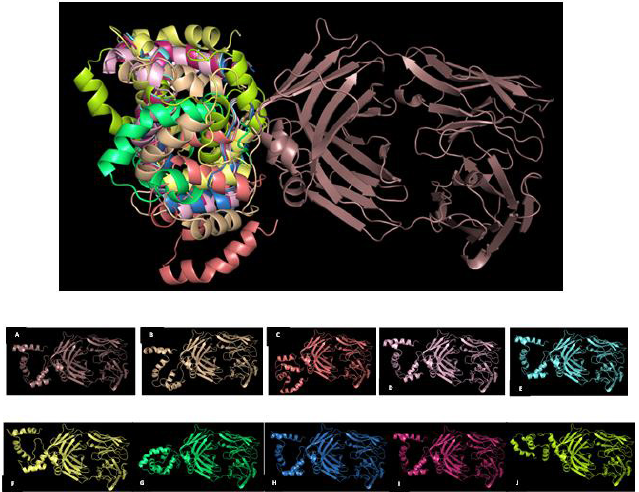
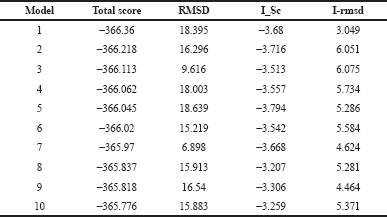
Generation of models ensemble in the server was conducted by 1,000 independent simulations in local docking perturbation run. Molecular docking had been run in the GrayLab. Rosetta-4 daemon and complete in 30 minutes. As shown in Figure 5, mostly all models (TLR4 with putative multiepitope) have similar conformation except model 3 (Fig. 5 C), model 7 (Fig. 5 G), and model 10 (Fig. 5 J). From 10 models, the best model score is model 1 with a total score of −366.36, RMSD value is 18.395, the interface score (I_Sc) value is −3.68, and I_rmsd value is 3.049 (Table 2). The quality of docking was categorized based on the accuracy of the closest decoy in the top five scoring decoys based on CAPRI-defined criteria: high (I_rmsd < 1.0 Å), medium (1.0 Å < I_rmsd < 2.0 Å), and acceptable (2.0 Å < I_rmsd < 4.0 Å) (Méndez et al., 2003). I_rmsd or interface rmsd is calculated over all backbone atoms in interface residues with an intermolecular distance of at most 4.0 Å (Chaudhury et al., 2011).
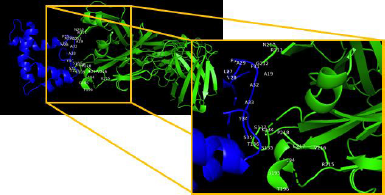
TLR4 has an active binding site as a favorable site of the multiepitope vaccine. TLR4 has two main chains such as I and J. Chain I represents Fab Hu 15C1 Heavy chain with a sequence length of 225 amino acid and no mutation. Chain J represents Fab Hu 15C1 Light chain with a sequence length of 214 amino acid and no mutation. Active binding sites favorable of multiepitope vaccine of TLR4 Chain I are S137, K134, T136, S135, P218, E217, V216, R215, G195, T196, and L194 and of Chain J are N210, R211, and G212. Amino acid residues in multiepitope that have interaction with active binding site in TLR4 are A19, L27, V28, A29, P30, F31, A32, A33, Y34, and S35 (Fig. 6).
Molecular dynamic simulation of TLR4 and putative multiepitope vaccine
The result of the molecular dynamic simulation is diversity, consisting of RMSD, Rg, Fraction of Native Contacts (Q), RMSF, B-factor, principal component analysis (PCA), analysis of dynamic cross-correlations, analysis of hydrogen bond, and energy calculation.
Molecular dynamics (MD) simulations of single-molecule and molecular complexes, including unbiased and biased MD simulations, are widely applied to achieve accurate binding modes, binding energies of drug-receptor interactions, drug-target recognition and binding, and allosteric mechanism research. MD simulations have been successfully applied in many drug discovery researches. The information from the dynamic trajectory could be used in determining the relationship between protein structure and function.
RMSD refers to the measure of the average distance between the atoms of superimposed structures. Sometimes, equalized RMSD plots indicate the equilibrium of the system.
Based on the RMSD graph shown in Figure 7 A, during molecular dynamic simulation record per frame time (ps) in X-axis, RMSD score overall is stable from 500 ps with a range of RMSD 0.0820–2.8577 Å. Based on RMSD histogram of the receptor shown in Figure 7 B, RMSD value range starts from 2.0 to 2.8 Å with the highest number in value around 2.5 Å. Overall spectrum of RMSD did not show any shifts which explains structure stability and strength of vaccine inside the active site pocket.
 | Figure 5. Overlapping visualization of 10 best models from molecular docking TLR4 with putative multiepitope vaccine; (A) Model 1; (B) Model 2; (C) Model 3; (D) Model 4; (E) Model 5; (F) Model 6; (G) Model 7; (H) Model 8; (I) Model 9; (J) Model 10. [Click here to view] |
 | Table 2. Docking energy of putative multiepitope vaccine with TLR4. [Click here to view] |
 | Figure 6. Amino acid interaction of TLR4 with putative multiepitope vaccines. [Click here to view] |
Rg or gyradius is used to characterize the dynamic trajectory of flexible systems for molecular systems. The calculation of Rg is one of the important indicators used in predicting the structural activity of a macromolecule. The Rg is influenced by the changes in the folding state of the protein. This provides an important probe of the equilibrium unfolding reaction. A small Rg score indicates that throughout trajectory the proteins were in folded structure.
Based on the fraction of native contact analysis shown in Figure 8 A, Q(X) value during molecular dynamic simulation per time (ps) was decreased f/rom 0.999999165535 to 0.945987820625. The Rg plot in Figure 8 B shows structural activity of a macromolecule during 1,000 time (ps) with a range value of 28.2–29.4.
Nonnative contacts have no significant part in the mechanism of folding in most cases. Coarse-grained theoretical and simulation models of folding support that only native contacts are energetically favorable. The fraction of Native Contacts Q(x) captured the transition sites of proteins with a folding free energy barrier. Q(x) will change with the unfolding of the protein.
RMSF refers to the atomic positional fluctuation. Here, the fluctuation of each residue is calculated based on the CA atom of them. The residue number in the crystal structure can be got from raw data at each residue position. At the bottom and top of the graphic, the secondary structure is indicated with colored lines (helices as black, strands as gray, and loops as white). During the simulation, these helices, strands, and loops are oriented to accommodate molecular interactions and ensure that the vaccine stays within the active site. Note that larger fluctuations are predicted for loop regions.
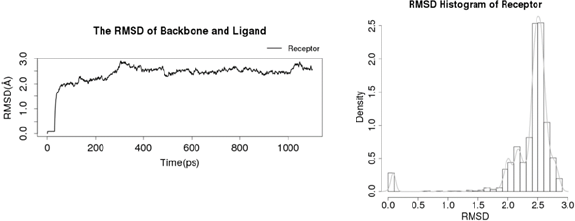 | Figure 7. (A) RMSD of protein backbone and ligand and (B) RMSD histogram of receptor. [Click here to view] |
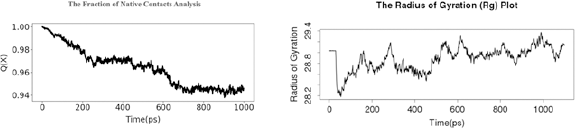 | Figure 8. (A) The fraction of native contact analysis and (B) the Rg. [Click here to view] |
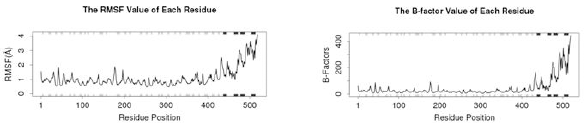 | Figure 9. (A) RMSF and (B) The B-factor. [Click here to view] |
B-factor, or the temperature factor, is similar to RMSF, which is used to describe atomic positional fluctuations, attenuation of X-ray scattering, or coherent neutron scattering caused by thermal motion. The residue number in the crystal structure can be got from raw data at each residue position. Similar to RMSF, at the bottom and top of the graphic, the secondary structure is indicated with colored lines. Note the larger fluctuations predicted for loop regions.
RMSF value of each residue (Fig. 9 A) shows a high fluctuation in residue position 430–500 and a low fluctuation in residue position 1–300. Similar to the RMSF value, the B-factor also has a low fluctuation in residue position 1–400 and has a high fluctuation in residue position 410–500 (Fig. 9 B).
PCA can be utilized to analyze the relationship between different structures based on their equivalent residues and the protein trajectories. The MD trajectory analysis vignette is used to discuss the application of PCA to visualize and compare the distribution of experimental structures and MD trajectories during the simulation. Studies suggest that 3–5 dimensions are sufficient for capturing more than 70% of the total variance in MD trajectories. The eigenvalue, variance, and cumulative of the top 6 PCs can be downloaded from the raw data for PC. PCA results showed in trajectory frames which colored from blue to red in chronological order. Because there are nearly only two statues for a protein, active and inactive, the frames are divided into two clusters in this server based on the top 3 PCs (Figs. 10A and B).
The atomic fluctuations of a system are correlated with one another and can be assessed by analyzing the magnitude of all pairwise cross-correlation coefficients. Blue indicates the correlated residues and red indicates the anticorrelated residues. The pairwise residues with the higher correlated coefficients (>0.8) and with a higher anticorrelated coefficient (<−0.4) are linked with light pink and light blue lines. The secondary structure schematic is added to the top and right margins of the dynamical residue cross-correlation map (helices black, strands gray, and loops white) (Fig. 11).
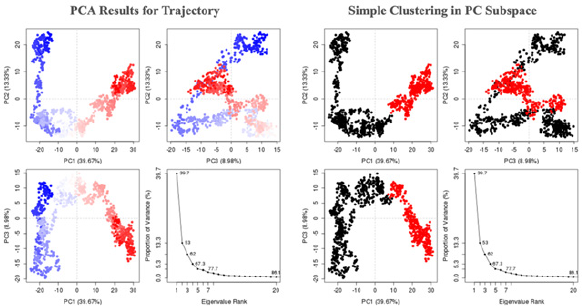 | Figure 10. (A) PCA results for trajectory and (B) simple clustering in PC subspace. [Click here to view] |
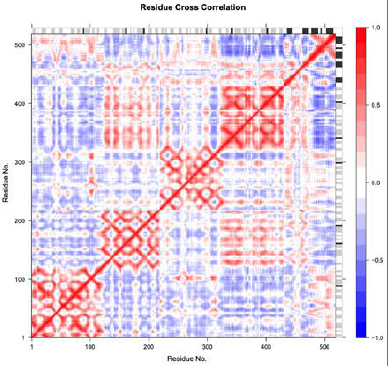 | Figure 11. Residue cross correlation. [Click here to view] |
CONCLUSION
In this study, we applied the immunoinformatics approach with genome diversity and conservancy analysis, in silico epitope prediction, molecular docking, and MD simulation in epitope-based breast cancer vaccine research. We identified seven putative epitope candidates from the conserved region of ERBB2, PTEN, and MUC4 protein, constructed as multiepitope which has the potential to be a new vaccine candidate for breast cancer. This approach may be considered as a new, safe, and efficient approach in vaccine research. Further research studies through in vitro and in vivo tests are necessary to validate the results.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
This research was supported by Penelitian Dasar Unggulan Perguruan Tinggi (PDUPT) Research Grant from the Ministry of Research and Technology of the Republic of Indonesia with Grants no. 8/E1/KP.PTNBH/2020 and 255/PKS/R/UI/2020.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This is a computational study and does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Andre I, Strauss CEM, Kaplan DB, Bradley P, Baker D. Emergence of symmetry in homooligomeric biological assemblies. Proc Natl Acad Sci, 2008; 105(42):16148–52. CrossRef
Atapour A, Negahdaripour M, Ghasemi Y, Razmjuee D, Savardashtaki A, Mousavi SM, Hashemi SA, Aliabadi A, Nezafat N. In silico designing a candidate vaccine against breast cancer. Int J Pept Res Ther, 2020; 26(1):369–80. CrossRef
Bateman, A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res, 2019; 47(D1):D506–15. CrossRef
Bazzichetto C, Conciatori F, Pallocca M, Falcone I, Fanciulli M, Cognetti F, Milella M, Ciuffreda L. PTEN as a prognostic/predictive biomarker in cancer: an unfulfilled promise? Cancers, 2019; 11(4):435. CrossRef
Benedetti R, Dell’Aversana C, Giorgio C, Astorri R, Altucci L. Breast cancer vaccines: new insights. Front Endocrinol, 2017; 8:270. CrossRef
Best RB, Hummer G, Eaton WA. Native contacts determine protein folding mechanisms in atomistic simulations. Proc Natl Acad Sci, 2013; 110(44): 17874–9. CrossRef
Bhasin M. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci, 2004; 13(3):596–607. CrossRef
Bocik WE, Sircar A, Gray JJ, Tolman JR. Mechanism of polyubiquitin chain recognition by the human ubiquitin conjugating enzyme Ube2g2. J Biol Chem, 2011; 286(5):3981–91. CrossRef
Bourne P, Weissig H. Structural bioinformatics. Wiley-Liss Inc, New York, NY, pp 321–7, 2003. CrossRef
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, 2018; 68(6):394–424. CrossRef
Brown TA, Mittendorf EA, Hale DF, Myers JW, Peace KM, Jackson DO, Greene JM, Vreeland TJ, Clifton GT, Ardavanis A, Litton JK, Shumway NM, Symanowski J, Murray JL, Ponniah S, Anastasopoulou EA, Pistamaltzian NF, Baxevanis CN, Perez SA, Papamichail M, Peoples GE. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res Treat, 2020; 181(2):391–401. CrossRef
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics, 2006; 7:153. CrossRef
Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, KeÅŸmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol, 2013; 9(10):e1003266. CrossRef
Case D, Betz R, Cerutti D, Cheatham ITE, Darden T, Duke R, Giese T, Gohlke H, Goetz A, Homeyer N. AMBER 2016 manual. University of California, Oakland, CA, 2016.
Chaudhury S, & Gray JJ. Identification of structural mechanisms of HIV-1 protease specificity using computational peptide docking: implications for drug resistance. Structure, 2009; 17(12):1636–48. CrossRef
Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One, 2011; 6(8):e22477. CrossRef
Chentoufi AA, Nesburn AB, BenMohamed L. Recent advances in multivalent self adjuvanting glycolipopeptide vaccine strategies against breast cancer. Arch Immunol Ther Exp, 2009; 57(6):409–23. CrossRef
Cherkasov A. Bioinformatics: a practical guide to the analysis of genes and proteins. Edited by Andreas D. Baxevanis and B. F. Francis Ouellette. ChemBioChem, 2005; 6(6):1128–9. CrossRef
Choudhuri, S. Bioinformatics for beginners: genes, genomes, molecular evolution, databases. Academic Press, Cambridge, MA, 2014. CrossRef
Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol, 2012; 8(10):e1002708. CrossRef
Delano WL. PyMOL : an open-source molecular graphics tool. DeLano Scientific, San Carlos, CA, 2002.
Dimitrov I, Flower DR, Doytchinova I. AllerTOP - a server for in silico prediction of allergens. BMC Bioinformatics, 2013; 14(Suppl 6):S4. CrossRef
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics, 2007; 8(1):4. CrossRef
Ferlay J, Ervik, M, Lam F, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F. Global cancer observatory.International Agency for Research on Cancer, Lyon, France, 2020. Available via https://gco.iarc.fr
Ghoncheh M, Pournamdar Z, Salehiniya, H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev, 2016; 17:43–6. CrossRef
Gopalakrishnan K, Sowmiya G, Sheik SS, Sekar K. Ramachandran plot on the web (2.0). Protein Pept Lett, 2007; 14(7):669–71. CrossRef
Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics, 2006; 22(21):2695–6. CrossRef
Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe, 2020; 27(4):671–80. CrossRef
Guex N, & Peitsch MC. SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modeling. Electrophoresis, 1997; 18(15):2714–23. CrossRef
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS. In silico approach for predicting toxicity of peptides and proteins. PLoS One, 2013; 8(9):e73957. CrossRef
Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids, 1999; 41:95–8.
Hattrup CL, Gendler SJ. Structure and function of the cell surface (Tethered) mucins. Annu Rev Physiol, 2008; 70(1):431–57. CrossRef
Johansson MU, Zoete V, Michielin O, Guex N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinformatics, 2012; 13:173. CrossRef
Joshi A, Joshi BC, Mannan MA, Kaushik V. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Inform Med Unlocked, 2020; 19:100338. CrossRef
Kalita P, Padhi AK, Zhang KYJ, Tripathi T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb Pathog, 2020; 145:104236. CrossRef
Kanampalliwar AM, Soni R, Girdhar A, Tiwari A. Reverse Vaccinology: Basics and Applications. J Vaccines Vaccin, 2013; 4:6.
Khan AM, Hu Y, Miotto O, Thevasagayam NM, Sukumaran R, Abd Raman HS, Brusic V, Tan TW, Thomas August J. Analysis of viral diversity for vaccine target discovery. BMC Med Genomics, 2017; 10(S4):78. CrossRef
Khan AM, Miotto O, Heiny AT, Salmon J, Srinivasan KN, Nascimento EJM, Marques ETA, Brusic V, Tan TW, August JT. A systematic bioinformatics approach for selection of epitope- based vaccine targets. Cell Immunol, 2006; 244(2):141–7. CrossRef
Khan AM, Miotto O, Nascimento EJM, Srinivasan KN, Heiny AT, Zhang GL, Marques ET, Tan, TW, Brusic V, Salmon J, August JT. Conservation and variability of dengue virus proteins: implications for vaccine design. PLoS Negl Trop Dis, 2008; 2(8):e272. CrossRef
Klein JS, Jiang S, Galimidi RP, Keeffe JR, Bjorkman PJ. Design and characterization of structured protein linkers with differing flexibilities. Protein Eng Des Sel, 2014; 27(10):325–30. CrossRef
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics, 2007; 23:2947–8. CrossRef
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics, 2007; 8(1):424. CrossRef
Li J, Zhang L, Li H, Ping Y, Xu Q, Wang R, Tan R, Wang Z, Liu B, Wang, Y. Integrated entropy-based approach for analyzing exons and introns in DNA sequences. BMC Bioinformatics, 2019; 20(S8):283. CrossRef
Loyau J, Didelot G, Malinge P, Ravn U, Magistrelli G, Depoisier JF, Pontini G, Poitevin Y, Kosco-Vilbois M, Fischer N, Thore S, Rousseau F. Robust antibody–antigen complexes prediction generated by combining sequence analyses, mutagenesis, in vitro evolution, x-ray crystallography and in silico docking. J Mol Biol, 2015; 427(16):2647–62. CrossRef
Ludovini V, Gori S, Colozza M, Pistola L, Rulli E, Floriani I, Pacifico E, Tofanetti FR, Sidoni A, Basurto C, Rull A, Crinò L. Evaluation of serum HER2 extracellular domain in early breast cancer patients: correlation with clinicopathological parameters and survival. Ann Oncol, 2008; 19(5):883–90. CrossRef
Lyskov S, & Gray JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res, 2008; 36:W233–8. CrossRef
Lyskov S, Chou FC, Conchúir SÓ, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, Borgo B, Havranek JJ, Kuhlman B, Kortemme T, Bonneau R, Gray JJ, Das R. Serverification of molecular modeling applications: the rosetta online server that includes everyone (ROSIE). PLoS One, 2013; 8(5):e63906. CrossRef
McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, Pande VS. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J, 2015; 109(8):1528–32. CrossRef
Méndez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins Struct Funct Bioinformatics, 2003; 52(1):51–67. CrossRef
Michel-Todó L, Reche PA, Bigey P, Pinazo MJ, Gascón J, Alonso-Padilla J. In silico design of an epitope-based vaccine ensemble for chagas disease. Front Immunol, 2019; 10:2698. CrossRef
Pallerla S, Abdul AURM, Comeau J, Jois S. Cancer vaccines, treatment of the future: with emphasis on her2-positive breast cancer. Int J Mol Sci, 2021; 22(2):1–16. CrossRef
Parvizpour S, Razmara J, Omidi Y. Breast cancer vaccination comes to age: impacts of bioinformatics. BioImpacts, 2018; 8(3):223–35. CrossRef
Patronov A, Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open Biol, 2013; 3:120139. CrossRef
Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov, 2007; 6(5):404–14. CrossRef
Raman H, Tan S, Sjaugi M, Mohamed N, Khan A. Bioinformatics for vaccine target discovery. Asia Pac Biotech News, 2014; 18(09):S0219030314001086.
Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins Struct Funct Bioinformatics, 2010; 78(9):2029–40. CrossRef
Safavi A, Kefayat A, Abiri A, Mahdevar E, Behnia AH, Ghahremani F. In silico analysis of transmembrane protein 31 (TMEM31) antigen to design novel multiepitope peptide and DNA cancer vaccines against melanoma. Mol Immunol, 2019; 112: 93–102. CrossRef
Sakib MS, Islam MR, Hasan AKMM, Nabi AHMN. Prediction of epitope-based peptides for the utility of vaccine development from fusion and glycoprotein of nipah virus using in silico approach. Adv Bioinformatics, 2014; 2014:402492. CrossRef
Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS Lett, 2006; 580(12):2985–92. CrossRef
Sivasubramanian A, Chao G, Pressler HM, Wittrup KD, Gray JJ. Structural model of the mAb 806-EGFR complex using computational docking followed by computational and experimental mutagenesis. Structure, 2006; 14(3):401–14. CrossRef
Sivasubramanian A, Maynard JA, Gray JJ. Modeling the structure of mAb 14B7 bound to the anthrax protective antigen. Proteins Struct Funct Bioinformatics, 2007; 70(1):218–30. CrossRef
Tani K, Murphy WJ, Chertov O, Salcedo R, Koh CY, Utsunomiya I, Funakoshi S, Asai O, Herrmann SH, Wang JM, Kwak LW, Oppenheim JJ. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int Immunol, 2000; 12(5):691–700. CrossRef
Tao ZQ, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys, 2015; 72(2):333–8. CrossRef
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, Jupe SC, Kok CY, Noble K, Ponting L, Ramshaw CC, Rye CE, Speedy HE, Stefancsik R, Thompson SL, Wang S, Ward S, Campbell PJ, Forbes SA. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res, 2019; 47(D1):D941–7. CrossRef
TopuzoÄŸullari M, Acar T, Pelit Arayici P, Uçar B, UÄŸurel E, Abamor EÅž, ArasoÄŸlu T, Turgut-Balik D, Derman S. An insight into the epitope-based peptide vaccine design strategy and studies against covid-19. Turk J Biol, 2020; 44(Special issue 1):215–27. CrossRef
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res, 2018; 46(W1):W296–303. CrossRef
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics, 2009; 25(9):1189–91. CrossRef
Yang JF, Wang F, Chen YZ, Hao GF, Yang GF. LARMD: integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Brief Bioinform, 2020; 21(6):2206–18. CrossRef