
INTRODUCTION
Acquired immunodeficiency syndrome (AIDS) is a worldwide epidemical issue concerning health and socio-economic issues. HIV infection mainly involves the destruction of helper-T cells, macrophages, dendritic cells, and cellular components, which affects the immune defense by involving cell-mediated immunity (Wong et al., 2010). As per the World Health Organization, globally, in 2018, people living with HIV had reached 37.9 million (Sheet, Global HIV & AIDS statistics – 2019 fact, 2019). The use of multiple drug therapies, which is also well-known as highly active antiretroviral treatment, is deemed as a significant advancement in the field of HIV therapy to combat the associated complications and deaths. Protease inhibitors are among the popular class of drugs used to inhibit HIV-1 protease, owing to their significant responsibility in the progression of Gag and Gag–Pro–Pol polyproteins to discrete proteins required for viral growth (Kohl et al., 1988). Darunavir (DRV) is a second-generation non-peptide synthetic amprenavir (APV) analog. It contains a bis-tetrahydrofuranyl (bis-THF) and a sulfonamide isostere. DRV obstructs the cleavage of HIV-encrypted Gag–Pol proteins in viral-infested cells selectively, thereby averting the development of advanced infectious virus particles (McCoy, 2007). Structural features displayed two THF rings, whereas APV had only one making DRV more superior. DRV can adapt as per the altering shape of protease due to its molecular flexibility. The DRV monotherapy also showed efficacy similar to triple therapy. The bis-THF functional group’s presence is found to influence the antiretroviral activity and allows the drug interaction with the critical amino acids of the HIV protease (De Meyer et al., 2005; Koh et al., 2003; Surleraux et al., 2005). DRV, a Biopharmaceutical Classification System Class II drug (low aqueous solubility), has a log p of 2.15 and pKa of 13.59. However, DRV suffers from extensive metabolism by the Cytochrome P450 enzyme (CYP3A4). It is also a substrate of P-gp efflux transporters, eventually resulting in low oral bioavailability (37%) (Thommes et al., 2009). Hence, it requires higher doses to achieve therapeutic effectiveness, which may also exaggerate gastrointestinal side effects leading to patient discomfort and noncompliance. The rationale behind polymeric nanocarriers as the delivery system for DRV is to overcome the problems mentioned earlier. A carrier system like polymeric nanoparticles (NPs) can provide controlled drug release, bypass hepatic first-pass metabolism, and enhance drug uptake by passive targeting. Attributed to its reduced particle size, prolonged circulation in the blood may help circumvent the receptors’ saturation, resulting in the modulation of the pharmacokinetics’ parameters (das Neves et al., 2015). Mangesh Bhalekar et al., (2017) reported on DRV’s solid lipid NPs, but the authors did not report the in vivo studies (Bhalekar et al., 2017). In this study, an attempt was made to enhance the selected drug’s bioavailability by preparing NPs using poly(ε-caprolactone) (PCL) NPs with Tocopherol Polyethylene Glycol Succinate 1,000 (TPGS) as a stabilizer.
MATERIALS AND METHODS
Materials
DRV and verapamil hydrochloride were gift samples from Hetero Pharma Pvt. Ltd. PCL (Mol. wt. 14000) and d-alpha-tocopheryl polyethylene glycol succinate 1,000 were purchased from Sigma-Aldrich (India). All other chemicals were purchased from the local market. Analytical grade solvents and reagents were used in the experiment.
Fabrication of darunavir NPs
The solvent emulsification–evaporation method was adopted with a slight modification (Sampathi et al., 2016). Briefly, DRV was taken in 5 ml acetone containing PCL and subjected to sonication to obtain a clear solution. The polymer–drug solution was added dropwise to 10 ml of the stabilizer solution under overhead stirring at 1,800 rpm to get a primary emulsion. The emulsion was sonicated for 15 minutes at an amplitude of 40% (5 seconds on/off cycle) using a probe sonicator (Sonics & Materials, Inc., Vibra cell VCX 750). The resulting emulsion was further injected slowly into the stabilizer solution and placed beneath a magnetic stirrer at 1,800 rpm which continued stirring for the entire evaporation of the organic solvent.
Screening of stabilizer
DRV NPs were formulated by screening different stabilizers (electrostatic and steric stabilizers) at concentrations of 0.5% w/v and 1% w/v. Depending on the particle size and poly-dispersity index (PDI), the stabilizer imparting the smallest particle size was considered for further optimization and other studies (Quintanar-Guerrero et al., 1996; Rangaraj et al., 2019).
Effect of stabilizer concentration
The result of stabilizer concentrations was assessed by keeping the drug to polymer ratio constant while varying the TPGS concentrations (0.02, 0.1, 0.25, 0.5, and 0.75% w/v). The particle size and PDI were measured for the prepared formulations (Quintanar-Guerrero et al., 1996).
Effect of sonication time
Sonication time plays an essential role in particle size reduction, where ultrasonic waves breakdown the agglomerates. Hence, optimization of sonication time is considered for formulation development. The effect of sonication time was evaluated at different time intervals (5, 10, 15, and 20 minutes) while keeping drug to polymer ratio as 1:1, and an internal and external phase containing a stabilizer of 0.25% w/v as the constant parameter (Khuroo et al., 2018).
Optimum preparation method for darunavir-loaded PCL NPs
Briefly, DRV (100 mg) was taken in 5 ml acetone containing 300 mg of PCL and subjected to sonication (10 minutes) to obtain a clear solution. The polymer–drug solution was added dropwise to 10 ml of the stabilizer solution (0.5% w/v TPGS) under overhead stirring at 1,800 rpm to get a primary emulsion. The emulsion was sonicated for 15 minutes at an amplitude of 40% (5 seconds on/off cycle) using a probe sonicator (Sonics & Materials, Inc, Vibra cell VCX 750). The resulting emulsion was further injected slowly (15 minutes) to about 40 ml of the TPGS solution (0.5% w/v) and placed beneath a magnetic stirrer at 1,800 rpm which continued stirring for 4 hours until the entire evaporation of the organic solvent.
Optimization of the cryoprotectant type and its concentration
The nano-formulations were subjected to centrifugation at 1,00,000 rpm for 30 minutes by employing an ultracentrifuge (R-24, REMI Electro-Tech Ltd.). The collected pellet was washed thrice with distilled water and redispersed in the distilled water (approximately 3 ml), followed by freeze-drying (Freeze dryer, FD 5508, Skadi-Europe) to obtain a free-flowing powder. Trials were carried out to select the type and concentration of cryoprotectant (0.5%, 1.0%, and 1.5% w/v) (Abdelwahed et al., 2006; Lee et al., 2009) during freeze-drying to enhance the NPs stability. Physical examination, such as reconstitution ability, redispersibility index, detachment from the round bottom flask walls, collapseness and volume covered by the freeze-dried NPs was noted for the freeze-dried formulations (Fonte et al., 2016). The prepared freeze-dried nanosuspension was dispersed in Milli Q water, suitably diluted, and analyzed for particle size. The ability of the nanosuspension to get redispersed is calculated by redispersibility index by the following equation
where D0 and D are the mean particle size before and after lyophilization, respectively (Rangaraj et al., 2019).
Characterization and evaluation
Particle size, PDI, and zeta potential
Particle size and PDI of the DRV NPs were measured by the dynamic light scattering principle using a Malvern Zetasizer (Nano ZS 90, Malvern Instruments, UK). Polymeric NPs were diluted tenfold with double distilled water. Zeta potential indicates the stability of the products based on electrostatic repulsion between particles. The zeta potential of NPs was determined by immersing a palladium electrode in the suspension of NPs using a zetasizer (NanoZS, Malvern Instruments, UK) (Rangaraj et al., 2019).
Entrapment efficiency (EE), drug-loading capacity (DLC), and percentage yield (PY)
Freeze-dried DRV NPs containing 50 mg of the drug were taken in the acetonitrile and methanol mixture in the ratio of 1:1. The formulations were subjected to bath sonication for 10 minutes and kept aside for 12 hours for ensuring the complete removal of the entrapped drug. After centrifuging at 8,000 rpm for 15 minutes, the samples were evaluated for their drug content at 266 nm via UV-Visible Spectrophotometer (JASCO, V 650) post-suitable dilution (van Loosdrecht et al., 1987; Sharma et al., 2014). EE, DLC, and PY were calculated by the following equations:EE
Determination of surface morphology characteristics
Surface morphology was studied using Scanning electron microscopy (SEM) (Shimadzu, S-3700N) at an accelerating 10-kV energy. The external morphology of the NPs was checked at 60,000 × intensification. For SEM examination, NP samples were fixated on a two-sided carbon tape, which sputtered with Au-Pd film in the presence of Argon in high vacuum (Byun et al., 2011).
Fourier Transform Infrared spectroscopy (FTIR) spectroscopy
FTIR spectra of DRV, PCL, mannitol, physical mixture, and optimized DRV-loaded NPs were recorded on a Perkin-Elmer 1,600 spectrophotometer (V-650 Jasco) potassium bromide (KBr) compressed pellet technique. Accurately weighed quantities of the drug and lyophilized DRV NPs (5 mg) were admixed with 50 mg KBr and were further triturated to get an excellent powder. The powder was placed in the pellet die, and a reasonable force was applied to get a thin pellet. The pellet was fixed in a die and scanned for IR spectra ranges from 4,000 cm−1 to 400 cm−1 (Zanetti et al., 2019).
Differential scanning calorimetry (DSC)
DSC thermograms indicate the presence of a physicochemical interaction and are used to detect drug–excipient compatibility. DSC analysis of DRV, PCL, and freeze-dried DRV-loaded NPs was carried out using a differential scanning calorimeter (Shimadzu, DSC-60, Kyoto, Japan). A thermal rate of 20°C minutes−1 in the scale of 0°C–200°C in a nascent nitrogen environment supplied at a speed of 40 ml/minutes was used (Ramazani et al., 2018).
In vitro dissolution studies by dialysis bag method
DRV release from drug suspension (plain drug dispersed with 0.5% w/v sodium carboxymethylcellulose) and NPs in 0.1N HCl and phosphate buffer pH 6.8 was performed under sink conditions. The dialysis bag (cut-off = 12,000–14,000 Dalton) containing plain drug suspension (equivalent to 1.5 mg of the drug) and DRV NPs (1.5 mg of the drug equivalent) was suspended in 50 ml of 0.1N HCl at 37°C at a stirring speed of 100 rpm for the first 2 hours. Later the media was replaced with phosphate buffer (pH 6.8) with 0.5% w/v Tween 80 (Ilkar Erdagi and Yildiz, 2019). At prefixed period points, 2 ml of the sample was acquired and substituted with fresh media. A UV-Visible spectrophotometer estimated the amount of drug in the aliquots at λmax 266 nm. The study was carried out in triplicates to calculate the mean cumulative percentage of drug release.
In vitro everted sac method
In vitro everted sac technique was used to investigate the efficacy of DRV NPs to protect the drug from P-gp efflux transporters. The impact of TPGS, when used as a stabilizer to inhibit the efflux of DRV, was checked by comparing plain DRV suspension with DRV NPs (Sha and Fang, 2004). The study protocol remained accepted by the Institutional Animal Ethics Committee (IAEC) with protocol number NIP/02/2013/PE/61. Female Sprague Dawley (SD) rats with an average weight of 220–250 g were fasted overnight before the study. Briefly, the animals used in the study were randomly allocated into three groups (plain DRV suspension, DRV + NPs, and DRV-loaded NPs stabilized by TPGS), with each group consisting of three animals. Surgery was performed using anesthetic ether; the intestine was isolated and washed with ice cold saline (50 ml). A length of 3 cm of the intestine was everted inside out, filled with tyrode solution (1 ml), ligated, and placed in a conical flask containing 25 ml buffer with formulations: DRV suspension, DRV along with blank NPs, and DRV NPs stabilized with TPGS. After 2 hours, the intestine drug content was measured using RP-HPLC to assess the extent of P-gp efflux inhibition of DRV by TPGS (Ravi et al., 2015).
Stability studies
Stability studies were carried out for lyophilized DRV NPs for 1 month as per the reported protocols (Jiang et al., 2009). The formulations were stored in 10 ml screw-capped glass vials and placed in a stability chamber at various temperatures 25°C ± 2°C, 60% ± 5% RH, 40°C ± 2°C, 75% ± 5% RH. Changes concerning particle size, PDI, redispersibility index, and EE were analyzed.
Plasma sample analysis
A pharmacokinetic study was carried out on SD rats weighing 220–250 gm. All experiments were carried out in compliance with CPCSEA guidelines, NIPER-Hyd (1548/PO/1/11 CPCSEA). The study’s protocol design was accepted by the IAEC bearing protocol number NIP/02/2013/PE/61. Animals under study were incubated under controlled temperature (25°C ± 2°C) and humidity (50% ± 5% RH). Before the day of the experiment, animals fasted overnight with free access to water. Randomly animals were categorized into four groups, with six animals per group. The first group was administered with plain drug dispersion (a drug with 0.5% w/v sodium CMC). On the contrary, the second group received optimized DRV NPs. The third group received blank NPs and the fourth group served as a negative control (Normal saline). The dose administered in all the groups was 20 mg/kg, concerning their body weight. After oral dosing, blood samples (500 μl) were acquired from retro-orbital plexus at each predetermined interval (0.5, 1, 2, 3, 4, 5, 6, 8, 12, 24, 30, and 36 hours) and were subjected to centrifugation at 5,000 rpm (8 minutes) to separate plasma.
To each plasma sample (100 μl), acetonitrile (300 μl containing 20 μg/ml of verapamil hydrochloride as internal standard) was added to precipitate the proteins. After vortexing and centrifugation at 5,000 rpm (10 minutes), the supernatant was collected. The respective samples’ drug content was analyzed using RP-HPLC (Hypersil BDS C-18 column) at an isocratic flow (60:40 ratio of acetonitrile and 50 mM phosphate buffer, pH 3.5). A flow rate (1.2 ml/minutes) was selected. Accuracy and precision (intraday and interday) were validated for the bioanalytical method (Supplementary Table 1). The calibration curve for DRV concentration in plasma was prepared, ranging between 100 and 1,000 ng/ml. It was found to be linear, with a regression coefficient of r2 = 0.999. HPLC in rat plasma containing drug and internal standard is shown in Supplementary Figure 1. The data of the bioanalytical method validation is given in Supplementary Table 1. Different pharmacokinetics were determined via Phoenix software Winnonlin version 6.3 (Pharsight, Certara company, Raleigh, NC).
RESULTS AND DISCUSSION
Formulation and optimization of DRV NPs
The solvent emulsification and evaporation method was used for the formulation of DRV-loaded NPs. Drug and the polymer were solubilized in 5 ml of acetone and added to an aqueous system comprising a surfactant (TPGS) under stirring. The resultant emulsion was sonicated and diluted further with the same surfactant solution. Solvent diffusion in the aqueous phase resulted in polymer precipitation. NPs were obtained after organic solvent evaporation. NPs, because of their small size and higher surface energies, drive these systems to aggregation. Hence, a stabilizer is mandatory to prevent the merger and agglomeration of NPs during and after their formation. The stabilizer’s adsorption at the boundary prevents the coalescence and particle aggregation by reducing interfacial tension (Jiang et al., 2009). Screening with different stabilizers was carried out to investigate the best stabilizer for stabilizing NPs.
In this study, Tween 80, Pluronic F127®, polyvinylpyrrolidone (PVP) K-30, and TPGS (0.5% and 1% w/v) were screened to select the best stabilizer based on the particle size and PDI (Table 1). Surfactant is very important to stabilize and to prevent the agglomeration of the formed NPs (Snehalatha et al., 2008). TPGS, an anionic stabilizer, gave the smallest particle size and PDI compared to other stabilizers, which may be attributed to steric repulsion provided by TPGS on the surface of NPs, was supported by its negative zeta potential values (Mu and Feng, 2003). Hence, TPGS was selected for further optimization. The outcome of sonication time on the NPs was carried out at various time points (5, 10, 15, and 20 minutes). The smallest particle size was observed when the nanoparticle batch was subjected to 15 minutes sonication (Fig. 1). It may be due to the most significant reduction in particle aggregation, which might be due to reduced viscosity of the aqueous segment, causing the polymer’s efficient homogenization throughout the aqueous phase and a subsequent particle size reduction (Sathyamoorthy et al., 2017). However, when subjected to 20 minutes sonication, the particle size increased. This may be due to more incredible surface energy disrupting the stabilizer layer over the polymer surface, leading to agglomeration and increased particle size (Al-Kaysi et al., 2005; Pagar and Vavia, 2013). The drug to polymer ratio was changed concerning different stabilizer concentrations from 1:1 to 1:4 at 0.25%–0.75% of the stabilizer. The effect of the stabilizer type and concentration with different drug to polymer ratios on the NPs’ characteristics is shown in Table 2. It was noticed that an increase in stabilizer concentration decreased the particle size of the NPs. The reason may be attributed to an optimum amount of stabilizer sufficient to form a layer and stabilize the NPs’ electrostatically (Cooper and Harirforoosh, 2014).
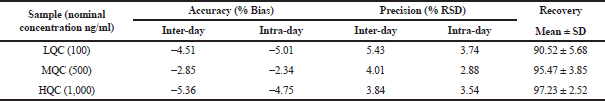 | Supplementary Table 1. Bioanalytical method validation. [Click here to view] |
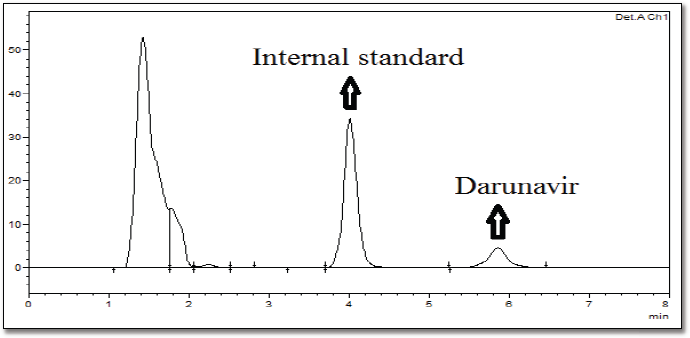 | Supplementary Figure 1. HPLC chromatogram of rat plasma containing drug and internal standard as additional data. [Click here to view] |
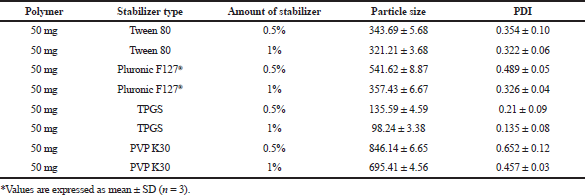 | Table 1. Stabilizer screening for the synthesis of NPs. [Click here to view] |
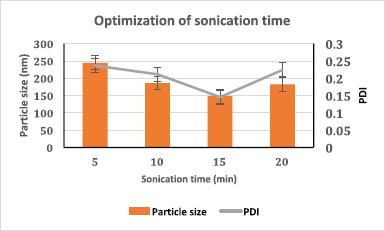 | Figure 1. Sonication time optimization based on particle size and PDI (n = 3, data represented as mean ± SD). [Click here to view] |
As shown in Table 2, an increased polymer concentration increased particle size, which may be due to stabilizer molecules’ insufficiency to stabilize the system by forming a layer over the polymer molecules, which is also reflected from the PDI results. While taking into consideration, PDI of the batches F1–F13 followed a similar trend in agreement with the particle size and PDI increased with an increased polymer concentration (Sharma et al., 2016). At the same time, a reduction in PDI is noted in conjunction with an increase in surfactant concentration. PDI gives an idea of dispersity and agglomeration of the particles in the system. An optimum amount of stabilizer and polymer results in reduced interfacial tension to give a more stable system.Zeta potential is directly dependent on the stabilizer concentration and indirectly on polymer concentration. Since TPGS is an anionic stabilizer, it imparts a negative charge on the surface of NPs providing stabilization via electrostatic repulsion (Fig. 2). The polymer PCL is neutral. An increase in TPGS amount increases zeta potential (Yang et al., 2018). However, a decrease in zeta potential was obtained with an increased drug to polymer ratio, which may be due to reduced molecules of stabilizer compared to the increasing polymer molecules, which thereby exerts a dilution effect and in turn reduces zeta potential (Ostolska and Wis̈niewska, 2014). Batch F1 had a particle size of 892.35 ± 3.3 nm, with a PDI of 0.427 ± 0.124, zeta potential of −1.68 ± 1.329 mV, and may be attributed to the absence of stabilizer. When other batches are taken into consideration, the particle size ranges from 99.2 ± 6.22 nm to 289.05 ± 6.99 nm with a change in PDI from 0.116 ± 0.038 to 0.269 ± 0.014 and zeta potential from −11.5 ±1.414 to −28.95 ± 0.494 mV.
Effect of cryoprotectant type and its concentration
Different cryoprotectants with varying concentrations were studied for their effect on the physical cakey appearance, collapseness, redispersibility index, detachment from the RB flask wall, and volume covered by the freeze-dried NPs. Based on the data obtained (Table 3), batch F8 containing mannitol (1% w/v concentration) was found to give the best results concerning its physical appearance, detachment from the wall, good reconstitution score, higher redispersibility index, and good volume recovery. Polysaccharide-based cryoprotectants were found to behave similarly as compared to PVP. The hierarchy of the cryoprotectants based on these parameters for PCL NPs leads us to conclude that Mannitol > Dextrose > Sucrose > PVP, respectively. Reduced mannitol percentage (M5) reduced the redispersibility index due to its insufficiency to form a monolayer over the NPs. When mannitol concentration increased, the redispersibility index was reduced. At high concentrations, the cryoprotectant disturbs the monolayer formation during the drying stage of post-freezing, thus leading to higher particulate growth with significant agglomeration on reconstitution (Abdelwahed et al., 2006).
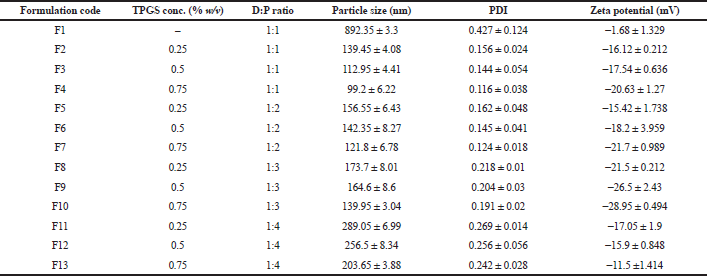 | Table 2. Mean particle size, PDI, and zeta potential of PCL NPs prepared by using different stabilizer concentrations along the drug to polymer ratio (n = 3). [Click here to view] |
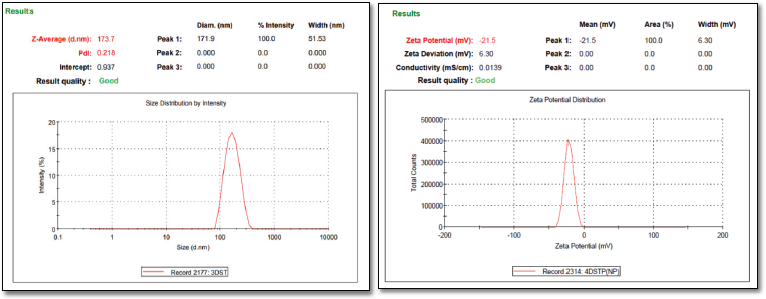 | Figure 2. (a) Particle size and PDI of DRV-loaded NPs (b) Zeta potential of DRV-loaded NPs. [Click here to view] |
EE, drug content, and PY
The EE of various formulations is depicted in Table 4. Drug content and EE are mainly associated with the drug to polymer ratio. An increase in polymer concentration resulted in an increased EE (Vuddanda et al., 2015) with maximum entrapment at a drug to polymer ratio of 1:3. However, a further increase in the polymer (a drug to polymer ratio of 1:4) slightly reduced the entrapment. Higher entrapment with the increasing polymer may be due to the drug’s low aqueous solubility, which acts as the driving force once the organic solvent starts to evaporate gradually. However, a further increase in polymer concentration led to a higher viscosity, leading to reduced diffusion velocity from the aqueous phase to the polymeric phase, leading to decreased EE. EE decreased with a subsequent rise in TPGS concentration in all drug to polymer ratios. The reduction in particle size was credited to the decreased entrapment. There is an increased tendency of drug diffusing from the organic phase to the external phase by reducing the surface tension with an increasing amount of TPGS, thus preventing drug entry into the polymeric matrix (Jeffery et al., 1993; Musumeci et al., 2006; Sharma et al., 2016).
EE and drug loading are indirectly dependent on the stabilizer concentration, decreasing with an increase in the stabilizer. This is due to reduced interfacial tension and energy differences between the two phases, thus preventing its diffusion into the polymer matrix.
Drug loading into the NPs depends on factors like the drug’s affinity toward polymer, physicochemical nature of the drug, polymer, type of the surfactant employed, and the method used to produce NPs (Panyam et al., 2004). An increase in polymer concentration increased the yield. However, an increment in the polymer concentration post-optimized batch resulted in reduced yield, which may be due to increased supersaturation, leading to a faster precipitation rate, preventing a proper formation of DRV-loaded spherical NPs (Table 4). Based on the size, zeta potential EE, drug loading, and %yield, formulation (F8) was selected for further studies. The particle size and zeta potential image of the same is shown in Figure 2.
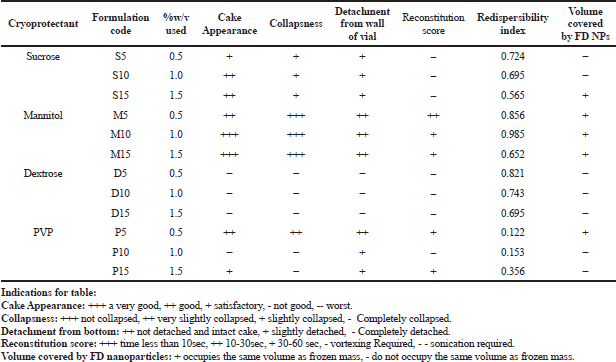 | Table 3. Studies on the effect of different cryoprotectants and concentration on lyophilized NPs formulation. [Click here to view] |
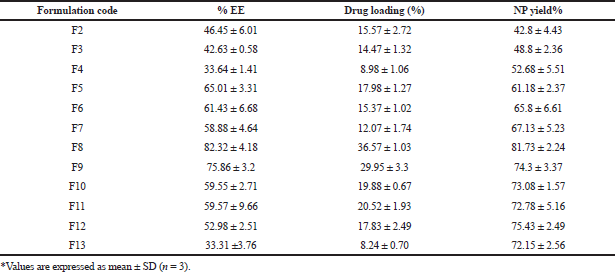 | Table 4. EE, drug loading, and yield of PCL NPs. [Click here to view] |
SEM analysis
SEM images of the optimized formulation are shown in Figure 3, indicating uniformly distributed spherical monodisperse particles. Smooth surface morphology with no particle aggregation was noted.
Fourier transform infrared spectroscopy (FTIR)
The compatibility between DRV and PCL was studied from the drug’s spectra, physical mixture, and freeze-dried NPs. DRV showed characteristic peaks (Fig. 4) at 3,464 cm−1 (OH group), 3,062 cm−1 (amine group), 3,028 cm−1 (aromatic C-H stretching), 2,916 cm−1 (methylene stretching), 1,709 cm−1 (carbonyl stretching), 1,598 cm−1 (aromatic C=C stretching), 1,239 cm−1 (CO stretching), aryl thio stretching at 700.16 cm−1. The appearance of similar peaks in the physical mixture and formulation indicated the compatibility between drug and excipients. The breakdown of NPs due to high pressure during the pellet preparation lead to the appearance of drug peaks in NPs (Desai and Thakkar, 2016).
 | Figure 3. SEM image of the DRV NPs. [Click here to view] |
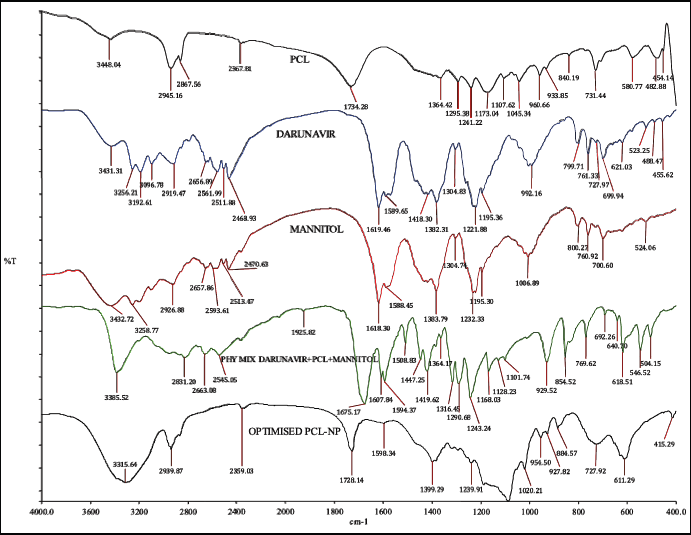 | Figure 4. IR overlay spectra of drug, polymer, mannitol, physical mixture & optimized DRV loaded NPs. [Click here to view] |
Differential scanning calorimetry (DSC)
DSC thermograms of the DRV, PCL, and freeze-dried NPs are shown in Figure 5. DRV exhibited a sharp endothermic peak at approx. 76.38°C. PCL and thermogram, depicted a sharp endothermic peak at 165.68°C, indicating a high degree of crystallinity. Drug peak was detected at 75.65°C in nanoparticle formulation matching the melting point of the DRV with slight differences. This may be attributed to the polymer’s encapsulation or dilution by a large polymer quantity (Mei et al., 2009; Zhang et al., 2012).
In vitro dissolution studies by dialysis bag method
In vitro release profile of DRV from PCL NPs and the plain drug is shown in Figure 6. NPs resulted in biphasic initial higher release up to 12 hours, along with a sustained release of the drug up to 72 hours, while plain drug suspension showed 96.87% ± 2.23% release within 6 hours. Initial burst release was attributed to surface adsorption of hydrophobic DRV on NPs and the high specific area of NPs, followed by a slower release of the entrapped drug (Chen et al. 2012). DRV diffusion from the PCL matrix may be responsible for the sustained release of DRV from NPs (Wang et al., 2012). Data treatment for drug release using various models (zero-order, first-order, Higuchi, and Korsemeyer–Peppa’s equations) is given in Table 5. The release was fitted well in Higuchi (r2 = 0.9485), indicating a diffusion type of release created by the polymer matrix and Korsemeyer–Peppa’s model [r2 = 0.9777; release exponent (n) as 0.44] indicating Fickian diffusion-controlled DRV release from NPs diffusing from the polymer matrix core to the surface (Sathyamoorthy et al. 2017).
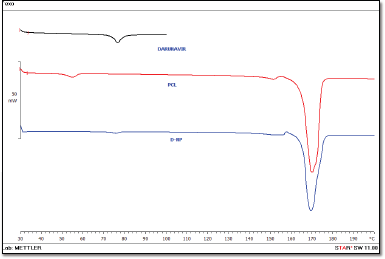 | Figure 5. DSC patterns of the DRV, PCL, and DRV-loaded NPs. [Click here to view] |
In vitro everted intestinal sac technique
The efficacy of the DRV NPs to prevent glycoprotein efflux was studied using the in vitro everted intestinal gut sac method as it is a simple and effective method (Cornaire et al., 2004). The everted gut sacs generally give better results as most carriers (P-gp receptors) predominantly exist on the mucosal side (Alam et al. 2012). The permeability results are shown in Figure 7. DRV permeability from the plain drug suspension inside the sac was significantly less at 120 minutes during the study period. This may be due to P-gp efflux of the drug from mucosal to the serosal wall, while drug absorption was similar (p > 0.05) when blank NPs along with DRV were studied. About threefold increase in permeability throughout ileum, duodenum, jejunum, and colon was noted when DRV encapsulated NPs were studied, which may be due to the presence of TPGS signifying its role as an efflux inhibitor. DRV encapsulation in nanosized carriers and P-gp efflux transporters’ inhibition by TPGS leads to increased transport of DRV across various intestinal barriers (Tavares Luiz et al. 2020; Yang et al., 2018).
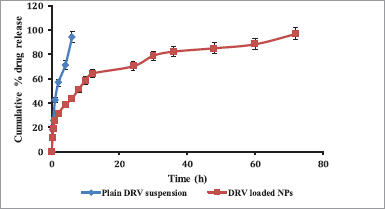 | Figure 6. In vitro release profile plain DRV suspension compared to DRV-loaded NPs (n = 3, data represented as mean ± SD). [Click here to view] |
 | Table 5. Release kinetics of DRV-loaded PCL NPs. [Click here to view] |
Stability studies
As indicated in Table 6, no significant (p < 0.05) changes in particle size, PDI, redispersibility index and entrapment were observed in the formulations when stored at 25 ± 2°C, 60 ± 5% RH, 40 ± 2°C, 75 ± 5% RH, and 2–8°C, respectively, indicating good stability of the formulation.
In vivo pharmacokinetic studies
The concentration-time profile of plain drug and NPs is shown in Figure 8. Pharmacokinetic parameters were estimated via Winnonlin software version 6.3 (Pharsight, Certara company, Raleigh, NC) and the data of the same is shown in Table 7. Significant improvement (p < 0.05) was observed in Cmax and AUC of DRV from formulation compared to a plain drug suspension. A 1.847-fold higher Cmax may be due to an increased permeation flux of DRV NPs than plain drug suspension. The elimination half-life of the drug in the form of DRV NPs increased by about 7 hours compared to a plain drug suspension. DRV-loaded NPs were successful in tremendously reducing the hepatic clearance of DRV compared to the plain drug. Mean residence time increased by twofold. AUC0–24 hours of DRV NP was found to be a 3.41-fold higher than plain DRV, indicating a significant improvement in bioavailability attributed to glycoprotein efflux inhibition by TPGS nanosized size and increased permeation by the polymeric carrier system (Kamaraj et al., 2017; Sampathi et al., 2016; Sonaje et al., 2007; Yu et al., 1999).
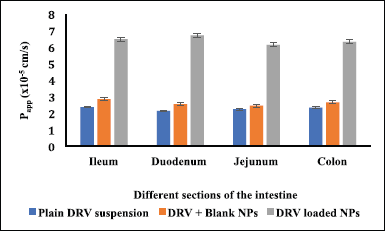 | Figure 7. Apparent permeability rate constant (Papp) of DRV across intestinal barriers using everted gut sac method (n = 3, data represented as mean ± SD). [Click here to view] |
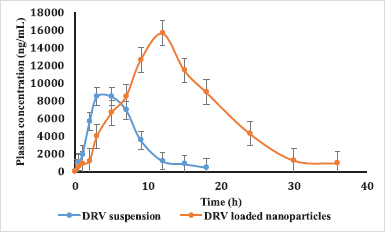 | Figure 8. Plasma concentration-time profiles of DRV in rats after oral administration of plain DRV suspension, DRV-marketed tablet, and DRV NP (50 mg/kg). Data represented as mean ± SD (n = 6). [Click here to view] |
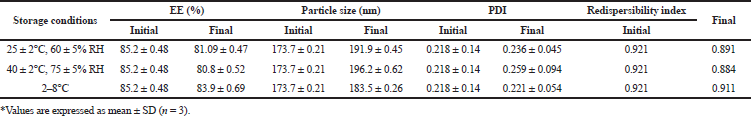 | Table 6. Stability study of optimized NPs. [Click here to view] |
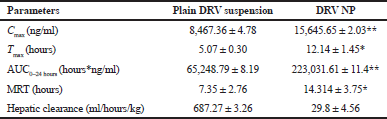 | Table 7. PK parameters after the oral administration of DRV suspension and DRV NP to rats (50 mg/kg, n = 3) *p < 0.05; **p < 0.01. [Click here to view] |
CONCLUSION
Polymeric NPs of DRV were formulated with solvent emulsification and evaporation method. The drug to polymer ratio and the polymer’s viscosity affected the following variables: particle size, EE, and PY of the NPs. Stability studies indicated that the prepared formulations were highly stable in particle size, PDI, EE, and redispersibility index. SEM depicted the formation of homogenous spherical NPs. The NPs also exhibited sustained release up to 72 hours. The everted sac technique confirmed the superiority of nanosized carriers and the efflux inhibitory activity of TPGS throughout the different sections of the intestine. Preparing NPs of DRV was thought to have a good potential in enhancing the drug’s systemic bioavailability in the presence of P-gp inhibitors like TPGS. A substantial decrease in hepatic clearance with an increase in MRT with a reduction of the first-pass metabolism and long plasma circulation time of the NPs increased oral bioavailability.
ACKNOWLEDGMENTS
The authors owe their gratitude to NIPER Hyderabad for providing facilities to carry out the present work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
The study protocol remained accepted by the Institutional Animal Ethics Committee (IAEC) with protocol number NIP/02/2013/PE/61. .
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Abdelwahed W, Degobert G, Fessi H. Investigation of nanocapsules stabilization by amorphous excipients during freeze-drying and storage. Eur J Pharm Biopharm, 2006; 63(2):87–94. CrossRef
Alam MA, Al-Jenoobi FI, Al-Mohizea AM. Everted gut sac model as a tool in pharmaceutical research: limitations and applications. J Pharm Pharmacol, 2012; 64:326–36. CrossRef
Al-Kaysi RO, Müller AM, Ahn TS, Lee S, Bardeen CJ. Effects of sonication on the size and crystallinity of stable zwitterionic organic nanoparticles formed by reprecipitation in water. Langmuir, 2005; 21(17):7990–94. CrossRef
Bhalekar M, Upadhaya P, Madgulkar A. Formulation and characterization of solid lipid nanoparticles for an anti-retroviral drug darunavir. Appl Nanosci (Switzerland), 2017; 7(1–2):47–57. CrossRef
Byun Y, Hwang JB, Bang SH, Darby D, Cooksey K, Dawson PL, Park HJ, Whiteside S. Formulation and characterization of α-tocopherol loaded poly e{open}-caprolactone (PCL) nanoparticles. LWT - Food Sci Technol, 2011; 44(1):24–8. CrossRef
Chen J, Li S, Shen Q. Folic acid and cell-penetrating peptide conjugated PLGA-PEG bifunctional nanoparticles for vincristine sulfate delivery. Eur J Pharm Sci, 2012; 47(2):430–43. CrossRef
Cooper DL, Harirforoosh S. Effect of formulation variables on preparation of celecoxib loaded polylactide-co-glycolide nanoparticles. PLoS One, 2014: 9(12):e113558. CrossRef
Cornaire G, Woodley J, Hermann P, Cloarec A, Arellano C, Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm, 2004; 278(1):119–31. CrossRef
das Neves J, Nunes R, Machado A, Sarmento B. Polymer-based nanocarriers for vaginal drug delivery. Adv Drug Deliv Rev, 2015; 92:53–70. CrossRef
De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, De Béthune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother, 2005; 49(6):2314–21. CrossRef
Desai J, Thakkar H. Effect of particle size on oral bioavailability of darunavir-loaded solid lipid nanoparticles. J Microencapsul, 2016; 33(7):669–78. CrossRef
Fonte P, Reis S, Sarmento B. Facts and evidences on the lyophilization of polymeric nanoparticles for drug delivery. J Control Release, 2016; 225:75–86. CrossRef
Ilkar Erdagi S, Yildiz U. Diosgenin-conjugated PCL-MPEG polymeric nanoparticles for the co-delivery of anticancer drugs: design, optimization, in vitro drug release and evaluation of anticancer activity. New J Chem, 2019; 43(17):6622–35. CrossRef
Jeffery H, Davis SS, O’Hagan DT. The preparation and characterization of poly(lactide-co-glycolide) microparticles. II. The entrapment of a model protein using a (Water-in-Oil)-in-Water emulsion solvent evaporation technique. Pharm Res, 1993; 10(3):362–68. CrossRef
Jiang J, Oberdörster G, Biswas P. Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies. J Nanoparticle Res, 2009; 11(1):77–89. CrossRef
Kamaraj N, Sujatha S, Alwin D. Dda loaded pcl nanoparticles enhances the oral bioavailability of dda in diabetes induced experimental rats. Int J Pharm Pharm Sci, 2017; 9(4):198. CrossRef
Khuroo T, Verma D, Khuroo A, Ali A, Iqbal Z. Simultaneous delivery of paclitaxel and erlotinib from dual drug loaded PLGA nanoparticles: Formulation development, thorough optimization and in vitro release. J Mol Liq, 2018; 257:52–68. CrossRef
Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother, 2003; 47(10):3123–9. CrossRef
Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RAF, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci U S A, 1988; 85(13):4686–90. CrossRef
Lee MK, Kim MY, Kim S, Lee J. Cryoprotectants for freeze drying of drug nano-suspensions: effect of freezing rate. J Pharm Sci, 2009; 98(12):4808–17. CrossRef
McCoy C. Darunavir: a nonpeptidic antiretroviral protease inhibitor. Clin Ther, 2007; 29(8):1559–76. CrossRef
Mei L, Zhang Y, Zheng Y, Tian G, Song C, Yang D, Chen H, Sun H, Tian Y, Liu K, Li Z, Huang L. A novel docetaxel-loaded poly (ε-caprolactone)/Pluronic F68 nanoparticle overcoming multidrug resistance for breast cancer treatment. Nanoscale Res Lett, 2009; 4(12):1530–9. CrossRef
Mu L, Feng SS. PLGA/TPGS nanoparticles for controlled release of paclitaxel: effects of the emulsifier and drug loading ratio. Pharm Res, 2003; 20(11):1864–72. CrossRef
Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, Puglisi G. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm, 2006; 325(1–2):172–9. CrossRef
Ostolska I, Wis̈niewska M. Application of the zeta potential measurements to explanation of colloidal Cr2O3stability mechanism in the presence of the ionic polyamino acids. Colloid Polym Sci, 2014; 292(10):2453–64. CrossRef
Pagar K, Vavia P. Rivastigmine-loaded L-lactide-depsipeptide polymeric nanoparticles: decisive formulation variable optimization. Sci Pharm, 2013; 81(3):865–85. CrossRef
Panyam J, William D, Dash A, Leslie-Pelecky D, Labhasetwar V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J Pharm Sci, 2004; 93(7):1804–14. CrossRef
Quintanar-Guerrero D, Fessi H, Allémann E, Doelker E. Influence of stabilizing agents and preparative variables on the formation of poly(D,L-lactic acid) nanoparticles by an emulsification-diffusion technique. Int J Pharm, 1996; 143(2):133–41. CrossRef
Ramazani A, Keramati M, Malvandi H, Danafar H, Kheiri Manjili H. Preparation and in vivo evaluation of anti-plasmodial properties of artemisinin-loaded PCL–PEG–PC nanoparticles. Pharm Dev Technol, 2018; 23(9):911–20. CrossRef
Rangaraj N, Pailla SR, Chowta P, Sampathi S. Fabrication of Ibrutinib nanosuspension by quality by design approach: intended for enhanced oral bioavailability and diminished fast fed variability. AAPS PharmSciTech, 2019; 20(8). CrossRef
Ravi PR, Vats R, Dalal V, Gadekar N, Aditya N. Design, optimization and evaluation of poly-ε-caprolactone (PCL) based polymeric nanoparticles for oral delivery of lopinavir. Drug Dev Ind Pharm, 2015; 41(1):131–40. CrossRef
Sampathi S, Achanta R, Tiriya P, Gera S, Reddy PS, Talla V. Formulation and evaluation of carvedilol loaded lactide nanoparticles: an appraisal to anti-hypertensive and anti-inflammatory therapy. Adv Sci Eng Med, 2016; 8(7):503–14. CrossRef
Sathyamoorthy N, Magharla D, Chintamaneni P, Vankayalu S. Optimization of paclitaxel loaded poly (ε-caprolactone) nanoparticles using Box Behnken design. Beni Suef Univ J Basic Appl Sci, 2017; 6(4):362–73. CrossRef
Sha X, Fang X. Transport characteristics of 9-nitrocamptothecin in the human intestinal cell line Caco-2 and everted gut sacs. Int J Pharma, 2004; 272(1–2):161–71. CrossRef
Sharma D, Maheshwari D, Philip G, Rana R, Bhatia S, Singh M, Gabrani R, Sharma SK, Ali J, Sharma RK, Dang S. Formulation and optimization of polymeric nanoparticles for intranasal delivery of lorazepam using Box-Behnken design: In vitro and in vivo evaluation. Biomed Res Int, 2014; 2014:1–12. CrossRef
Sharma N, Madan P, Lin S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: a co-surfactant study. Asian J Pharm Sci, 2016; 11(3):404–16. CrossRef
Sheet, Global HIV & AIDS statistics – 2019 fact. Unaids. 2019. Available via https://www.unaids.org/en/resources/fact-sheet 2019. (Accessed 11 December 2020).
Snehalatha M, Venugopal K, Saha RN, Babbar AK, Sharma RK. Etoposide loaded PLGA and PCL nanoparticles II: biodistribution and pharmacokinetics after radiolabeling with Tc-99m. Drug Deliv, 2008; 15(5):277–87. CrossRef
Sonaje K, Italia JL, Sharma G, Bhardwaj V, Tikoo K, Kumar MNVR. Development of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in rats. Pharm Res, 2007; 24(5):899–908. CrossRef
Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, De Kock, HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, De Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem, 2005; 48(6):1813–22. CrossRef
Tavares Luiz M, Delello Di Filippo L, Carolina Alves R, Sousa Araújo VH, Lobato Duarte J, Maldonado Marchetti J, Chorilli M. The use of TPGS in drug delivery systems to overcome biological barriers. Eur Polym J, 2020; 142:1–15. CrossRef
Thommes M, Baert L, van ‘t Klooster G, Geldof M, Schueller L, Rosier J, Kleinebudde P. Improved bioavailability of darunavir by use of κ-carrageenan versus microcrystalline cellulose as pelletisation aid. Eur J Pharm Biopharm, 2009; 72(3):614–20. CrossRef
van Loosdrecht MC, Lyklema J, Norde W, Schraa G, Zehnder AJ. Electrophoretic mobility and hydrophobicity as a measured to predict the initial steps of bacterial adhesion. Appl Env Antimicrob, 1987; 53(8):1898–901. CrossRef
Vuddanda PR, Mishra A, Singh SK, Singh S. Development of polymeric nanoparticles with highly entrapped herbal hydrophilic drug using nanoprecipitation technique: an approach of quality by design. Pharm Dev Technol, 2015; 20(5):579–87. CrossRef
Wang Y, Wang C, Gong C, Wang Y, Guo G, Luo F, Qian Z. Polysorbate 80 coated poly (ε-caprolactone)-poly (ethylene glycol)-poly (ε-caprolactone) micelles for paclitaxel delivery. Int J Pharm, 2012; 434(1–2):1–8. CrossRef
Wong HL, Chattopadhyay N, Wu XY, Bendayan R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv Drug Deliv Rev, 2010; 62:503–17. CrossRef
Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics, 2018; 8:464–85. CrossRef
Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A, Winnike R, Coffin M. Vitamin E-TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res, 1999; 16(12):1812–7. CrossRef
Zanetti M, Mazon LR, de Meneses AC, Silva LL, de Araújo PHH, Fiori MA, de Oliveira D. Encapsulation of geranyl cinnamate in polycaprolactone nanoparticles. Mater Sci Eng C, 2019; 97:198–207. CrossRef
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials, 2012; 33:4889–906. CrossRef
GRAPHICAL ABSTRACT
 | [Click here to view] |