
INTRODUCTION
Oral route of administration is the most frequently used route of drug administration and generally, the most convenient for the patient. For an oral drug to be effective, a therapeutic concentration in the blood should be reached. This concentration is mostly dependent on its bioavailability, which is affected primarily by the rate and extent of absorption. As Amidon et al. (1995) described back in 1995, oral absorption of a drug is vitally reliant on its ability to solubilize in an aqueous media and its intestinal permeability.
The Biopharmaceutical Classification System is an outline for classifying drug compounds based on their solubility and permeability (Yasir et al., 2010). Carvedilol belongs to class II and exhibits low aqueous solubility but have high permeability, thus any factor affecting the dissolution characteristics would have a reflective influence on the bioavailability.
Solubility is a critical factor in drug dissolution and further absorption, and therefore, it plays an important role in oral bioavailability. For a drug to be absorbed, it must first be present at the site of absorption in the form of an aqueous solution (Savjani et al., 2012). For class II drugs that can cross the intestinal mucosa easily, the lack of ability to go into solution is sometimes the limiting step to its overall bioavailability. Dissolution usually depends on the physicochemical characteristics of the drug such as partition coefficient, solubility, salt formation, surface area, particle shape, size, crystallinity, polymorphism, and stereo chemical factor (Custodio et al., 2008). A classical approach to enhance solubility without affecting its biological activity is by chemical modification, in which salts and prodrug are generated by incorporation of polar or ionizable moieties into the parent drug. A different strategy to solve solubility issues is by a formulation method that includes the use of co-solvents, surfactants, complexation agents, co-crystal formation, and solid dispersions (Kovvasu et al., 2018a; Zhang et al., 2012a).
Solid dispersions can be defined as dispersions of a drug in an amorphous polymer matrix or small molecule where the drug is preferably in the molecularly dispersed state (Huang and Dai, 2014). Solid dispersions have become an established technology to augment the solubility, dissolution, and bioavailability of poorly soluble drugs (Kovvasu et al., 2018b). Determining the proper matrix-drug ratio is of high importance since a higher drug loading will lead to spontaneous separation of phases followed by crystallization of the drug (Tian et al., 2015). Surface active agents are often used to improve drug loading providing other benefits such as decreased aggregation, improved wetting, and increased drug solubility. The addition of surface active agents to a solid dispersion have proven the ability to increase drug solubility, dissolution rate, and similarly enhance the oral bioavailability (Zhang et al., 2012b).
Carvedilol is a nonselective β-adrenergic blocking agent with selective α1-adrenergic blocking activity for the treatment of hypertension and congestive heart failure (Dunn et al., 1997). Carvedilol absolute bioavailability following oral administration is only ~25% because of its slow dissolution rate and extensive first pass metabolism (Lee, 2012). Carvedilol is also the substrate of P-glycoprotein, reducing its intestinal absorption by actively pumping it out of cells (Bart et al., 2005). Carvedilol exhibits low oral bioavailability due to its dissolution rate-limiting oral absorption, hence it was chosen as a model drug for this study. In the present investigation, we formulated a solid dispersion system comprised of a mixture of Gelucire 44/14 or Gelucire 50/13, nonionic water-dispersible surfactants for lipid-based formulations commonly used to solubilize and increase oral bioavailability of poorly water-soluble carvedilol. Dissolution studies were performed to characterize the dissolution profile of the various formulations. Subsequently, the effect of incorporating D-α-tocopheryl polyethylene glycol succinate (vitamin E TPGS/TPGS), also a nonionic surfactant, into the lead formulation to further enhance the dissolution of carvedilol was also investigated, a third generation solid dispersion formulation. Similarly, parallel artificial membrane permeability assay (PAMPA) studies to assess the enhanced permeation effect of the formulation were performed.
Stability studies were conducted on the optimized formulations by exposing them to accelerated (40°C/75% RH), real time (25°C/60% RH), and refrigerated (2°C–8°C) conditions for a period of 12 weeks. Last, the oral bioavailability of carvedilol solid dispersion was evaluated in vivo after administration to Sprague-Dawley rats. Though several studies are reported in the literature to improve the oral bioavailability of carvedilol, there have been no attempts to use a third generation solid dispersion system combining two different active surface carriers such as Gelucire and TPGS. We, therefore, attempted to prepare a solid dispersion of carvedilol to study the synergetic effect of Gelucire in the presence of TPGS.
MATERIALS AND METHODS
Materials
Drug substance carvedilol was obtained from TCI America (Portland, OR). Gelucire 40/14 and Gelucire 50/13 were gifted by Gattefossé (St. Priest, France). Vitamin E TPGS was purchased from Eastman Chemical Company (Anglesey, United Kingdom). Amorphous fumed silica and micro-crystalline cellulose (MCC) were procured from Alfa Aesar (Ward Hill, MA). Croscarmellose sodium was purchased from Spectrum Chemicals (New Brunswick, NJ). PAMPA 96-well plates were purchased from Millipore Sigma (Billerica, MA). Sprague-Dawley rats were procured from Harlan Incorporated (Indianapolis, IN) and Sprague-Dawley rat plasma was obtained from Valley Biomedical (Winchester, VA). All other materials and organic solvents were at least of HPLC grade and were purchased from Millipore (Billerica, MA).
Preparation of solid dispersions
In a first stage, various solid dispersions with different ratios of Gelucire 44/14 or Gelucire 50/13 and carvedilol were prepared by the fusion-solvent method. Briefly, selected Gelucire was melted at 60°C using a hot plate and carvedilol was incorporated to the melt. Then, ethanol was added to the mixture to ensure full solubilization of carvedilol evaluated as a clear solution. and then adsorbed onto a mixture of amorphous fumed silica and MCC (1:2) at a fixed ratio of 1 part of melted Gelucire for each 2.25 parts of solid carrier mixture to form a solid coarse powder. The resultant powder was subjected to vacuum desiccation to remove any solvent residues, pulverized, and passed through a 60-mesh sieve to obtain free-flowing powder. This powder was used to fill size 1 capsules or compressed to form tablets with a final dose equivalent to 25 mg of carvedilol. Kambert mini rotary tablet press (Kambert Machinery Co. Private Ltd., Ahmedabad, India) was used to form the tablets. Croscarmellose sodium (15% w/w) was used as tablet disintegrant. Second stage started after dissolution screening studies, in which a lead formulation was selected. To this formulation, TPGS was added in different percentages (w/w) to the melted Gelucire/carvedilol/ethanol mixture to investigate if incorporation of a second surface-active ingredient, and followed the same procedure of manufacturing as mentioned above, could potentially improve dissolution of the drug.
Analysis of carvedilol
For the analysis of carvedilol, a modified reversed-phase HPLC method was developed using Waters HPLC system (2487/600/717) coupled to an UV-visible detector (Waters, San Diego, CA). As mobile phase, 50 mM sodium dihydrogen phosphate (NaH2PO4) buffer with pH adjusted to 2.5 using diluted orthophosphoric acid and methanol in a fixed 40:60 (buffer:methanol, v/v) ratio were used. Flow rate was set to 1.0 ml/min with an injection volume of 10 μl. The chromatographic separation was done on a Phenomenex Luna C-18 (5 μm × 4.6 mm × 150 mm) and the elution was measured at 240 nm.
Calibration standards and quality control (QC) samples
Standard stock solutions (n = 3) were prepared by dissolving 25 mg of drug in 25 ml of methanol in 25-ml volumetric flask. Further dilutions with same solvent were prepared to obtain different working standard solutions ranging from 10–200 μg/ml, where 10 and 200 μg/ml are low and high concentrations.
Linearity and range
Linearity was assessed using freshly prepared standard solutions covering the range of 10–200 μg/ml. In addition, blank samples with blank mobile phase were also run to reject the presence of interfering peaks.
Precision and accuracy
For determination of intra-day precision, inter-day precision and accuracy, QC samples were analyzed both within a single run and between different runs. Method accuracy was calculated from the ratio of measured concentration to the nominal added concentration, based on the standard curve. Precision was evaluated by calculating the inter-day and intra-day variation by calculating the relative standard deviation of measured concentration (%RSD) over a period of 3 days.
Robustness
To determine the robustness of the method, flow rate was altered and evaluated for the effect of these changes on the signal response. Three different flow rates that include 0.8, 1.0, and 1.2 ml/minute were evaluated. %RSD was calculated for individual parameters to determine the robustness of the method.
Assay of carvedilol
To determine the assay of drug present in the powder formulation, the previously described HPLC method was used. Briefly, a precisely weighed portion of the sample powder, equivalent to about 40 mg of drug, was transferred to a 200-ml volumetric flask. Next, 150 ml of solvent were added and sonicated for 20 minutes. The flask was cooled down to room temperature and then diluted to volume. An aliquot of the above solution was centrifuged to remove impurities and supernatant was injected to the HPLC.
Differential scanning calorimetry
Differential scanning calorimetric (DSC) test runs were performed to assess the physical state of the drug in the different formulations. DSC profiles of carvedilol, carvedilol physical mixture, carvedilol solid dispersion, and excipients used were carried out using a DSC-823e (Mettler Toledo LLC, USA) connected to a FT-100 refrigeration unit (Julabo USA Inc, USA). Aluminum pans were used in the studies. Samples were carefully weighed in non-hermetically sealed aluminum pans with weights ranging between 5 and 10 mg. An empty pre-weight aluminum pan was used as reference. The sample furnace was under a constant stream of nitrogen. The pan was heated from −10°C to 200°C at a heating rate of 10°C/minute.
In vitro dissolution studies
A variation of the method reported in the U.S. Pharmacopeia for carvedilol was used since the recommended dissolution medium was found to be not suitable for discriminating the dissolution rate to support formulation development for this dosage form. Therefore, dissolution testing was carried out adding sample equivalent to 25 mg of carvedilol to 900 ml of phosphate buffer pH 6.8. The paddle speed was set to 75 rpm. Sampling aliquots of 5.0 ml were withdrawn at 15, 30, 45, 60, 120, and 240 minutes. Sampled volumes were not replenished and appropriate adjustments were made in the calculation to compensate the loss. After collection, aliquots were filtered using a 0.45-μm membrane nylon filter to remove insoluble impurities and diluted with same dissolution media, before being analyzed by HPLC method described above. Dissolution studies were performed in triplicates and cumulative drug released percentage from capsules or tablets were calculated.
Parallel artificial membrane permeability assay (PAMPA)
A 96-well filter plate was used as permeation donor while a 96-well bottom plate served as receiver. Phosphate buffer adjusted to pH 6.8 was used as receptor and donor buffer in the studies. A 1% solution (w/v) of high purity lecithin dissolved in dodecane was prepared. Five μl of the aforementioned solution was then added carefully into each well of the filter plate to function as an artificial membrane. A suspension of carvedilol pure drug or selected formulations with an equivalent concentration of 40 mg/ml of carvedilol was prepared. One hundred and fifty μl of test article suspension was added to each well of the donor plate instantly after the application of the artificial membrane and 300 μl of the pH 6.8 phosphate buffer were added to each well of the receptor plate. Onto this plate, the drug-filled donor plate was carefully placed on the top, making sure that the bottom of the artificial membrane was in contact with the buffer throughout the whole receptor plate. The donor-receptor assembly was incubated for 16 hours at room temperature and a cover lid was used to cover the plate to avoid any evaporation. After the incubation time, samples aliquots from the donor and the receptor component were collected and analyzed for carvedilol concentration by HPLC method. Finally, permeability values for the samples were calculated according to the following formula (Chen et al., 2008).
Where Pe is the permeability in cm/s. A is the effective filter area = 0.24 cm2 for this specific plates, VD is the donor well volume which is 0.15 ml, VA is the receptor well volume which is 0.3 ml, t is the incubation time (s), CA(t) is the drug concentration in receptor well at time t, CD(t) is the drug concentration in donor well at time t, and
Stability studies
Stability studies were performed on carvedilol solid dispersion capsules by exposing them to 40°C/75% RH, 25°C/60% RH and 2°C–8°C for a period of 3 months. Capsules were prepared by the method described previously and filled into HDPE bottles with 1.0 g of cotton. Samples were withdrawn at predetermined time intervals and evaluated for their assay content and dissolution profile.
Pharmacokinetic studies
Male Sprague-Dawley rats with a body weight between 225 and 250 g were purchased and pre-jugular vein cannulated and used in the study. The rats were randomly assigned to three groups with six rats per group and fasted overnight with water ad libitum prior to the experiment. Utilizing a needle for oral gavage, group 1 was administered a suspension of pure drug carvedilol at a dose of 40 mg/kg and group 2 rats were given a suspension of solid dispersion carvedilol-Gelucire 50/13 (SD8) in water at equal dose. Group 3 was given a suspension of solid dispersion SD8 + 10% TPGS in water equivalent to 40 mg/kg. Three hundred μl of blood were collected into a heparinized Eppendorf tubes from the jugular vein at 0.5, 1, 2, 4, 6, 8 and 24 hours after administration. Blood samples were centrifuged at 10,000 rpm/4°C for 10 minutes. The plasma was carefully separated and stored at −80°C until analysis. After collection of all the samples, the rats were sacrificed by a lethal dose of isoflurane. The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC), Western University of Health Sciences.
Preparation of calibration standards and quality control samples
For carvedilol, a stock solution 1,000 ng/ml in 50% methanol was prepared. Working solutions 10, 20, 50, 100, 200, 500, and 1,000 ng/ml were prepared. Quality control samples at low (20 ng/ml) and high (500 ng/ml) concentrations were prepared in a similar way. Plasma samples were spiked with propranolol used as internal standard (IS).
Plasma sample preparation
For the extraction of drug and their corresponding IS, a protein precipitation extraction method was employed. For this purpose, 300 μl of methanol was added to each tube containing 100 μl of plasma. The above mixture was vortexed for 1 minute and centrifuged for 10 minutes at 13,000 rpm and 4°C. The organic supernatant was transferred into an HPLC vial for analysis.
Chromatographic condition
The analytical column was Zorbax SB-C18 (5 μm, 2.1 × 150 mm) (Agilent, USA) with a protective guard column (12.5 × 2.1 mm). Acetonitrile and 0.1% formic acid (40:60, v/v) constituted the mobile phase. The column was kept at room temperature, the flow rate was kept at 0.3 ml/minute with an injection volume of 5 μl and a triple-quadrupole LC-MS mass spectrometer (API 3200, AB Sciex, Framingham, MA) detector was used.
Range and linearity
Range and linearity of the method were calculated using fresh samples prepared in blank rat plasma and the calibration curves consisted of seven non-zero standard levels with a range of 10–1,000 ng/ml. Also, a blank sample was analyzed to reject any interference peaks. The samples were extracted and analyzed as described previously.
Precision and accuracy
To determine within-run and between-run precision and accuracy, a set of three different standards at concentrations of 20, 100, and 500 ng/ml were analyzed using the method described above. The accuracy was calculated from the ratio of measured concentration to the theoretical concentration based on the standard curve. Precision of the method was calculated using the within-run and between-run variation coefficients of the standards at each concentration level (%CV).
Freeze-thaw stability
To determine the effect on analyte levels in plasma samples subjected to freezing temperatures, samples with concentration levels of 20, 100, and 500 ng/ml were prepared in rat plasma and store at −80°C until completely frozen. Samples were then thawed unassisted at room temperature and analyzed for drug concentration.
Statistical analysis
Statistical analysis of the dissolution profiles was carried out by two-way analysis of variance (ANOVA) and that of PAMPA was carried out by one-way ANOVA.
RESULTS
The enhancement of oral bioavailability of poorly water-soluble drugs remains a challenging aspect of drug delivery. Recent years, third generation solid dispersion use lipid-based amphiphilic carriers with solubilizing properties, such as Gelucire 44/14 or TPGS, to achieve the highest degree of bioavailability of poorly soluble drugs (Serajuddin, 1999). In this study, carvedilol solid dispersion was successfully prepared with different ratios of Gelucire and TPGS using fusion-solvent method.
Preparation of solid dispersions
Various solid dispersion systems were successfully prepared by fusion-solvent method using different amounts of Gelucire 44/14 or 50/13 in combination with carvedilol as described in Table 1. The melted mixture of Gelucire and drug resulted in a clear homogeneous preparation, creating a favorable system were carvedilol was fully solubilized. The mixtures were effectively turned into a solid by adding amorphous fumed silica/ MCC under constant stirring. This dry powder was then filled into capsules or mixed with 15% croscarmellose and compressed into tablets for testing.
Third generation solid dispersion systems incorporating two surface active ingredients were successfully prepared by melting Gelucire 50/13 and adding TPGS in different concentrations (0%–10%). No phase separation or precipitation was observed in the mixture. Incorporation of TPGS to the melted mixture ensures a uniform distribution in the formulation. These mixtures containing carvedilol were then turned into solid powder by the incorporation of amorphous fumed silica/MCC and filled into capsules or compressed.
Analytical method validation
The retention time of carvedilol peak was 4.76 minutes. Eight-point calibration curve for carvedilol was linear over concentration range of 10–200 μg/ml. The equation for the standard curve was y = 2909.4x + 99.475 (r2 = 0.9997). %RSD values for within run and between run samples were less than 2%. Accuracy of carvedilol in the quality control samples was detected in the range of 99%–101%. %RSD values for the samples prepared for evaluating the robustness of the method were also less than 2% indicating the method to be very robust. The results of the analytical method validation are shown in Table 2.
 | Table 1. Formulation composition of carvedilol solid dispersions. [Click here to view] |
Differential scanning calorimetry
The DSC thermograms performed for pure drug, excipients, physical mixture, and solid dispersion formulation are exhibited in Figure 1. The thermogram representing pure carvedilol displayed a sharp melting point peak at 118.54°C, which complies of that of reporting data (Pokharkar et al., 2006). Physical mixture presented a short and not-well define endothermic peak in the temperature range corresponding to that of carvedilol, probably due to the melted Gelucire partially dissolving the drug. Among the excipients presented in the formulation, Gelucire 50/13 resulted in a broad endothermic peak at 48.61°C, while TPGS revealed a sharp peak at 40.88°C. Thermogram of solid dispersion system presented no endothermic peak in the range previously observed by the carvedilol. This can be explained by a complete homogeneous dispersion within the carrier and conversion to amorphous state by overcoming crystal lattice energy. This change in the melting point of carvedilol as a solid dispersion might be due to the physical interactions between the excipients and the drug. Consequently, the enhanced solubility of carvedilol, as observed during the dissolution studies, might be attributed to transformation of crystalline state of drug to an amorphous state.
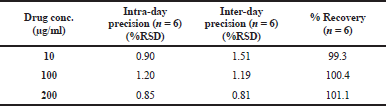 | Table 2. HPLC analytical method validation results for determination of carvedilol. [Click here to view] |
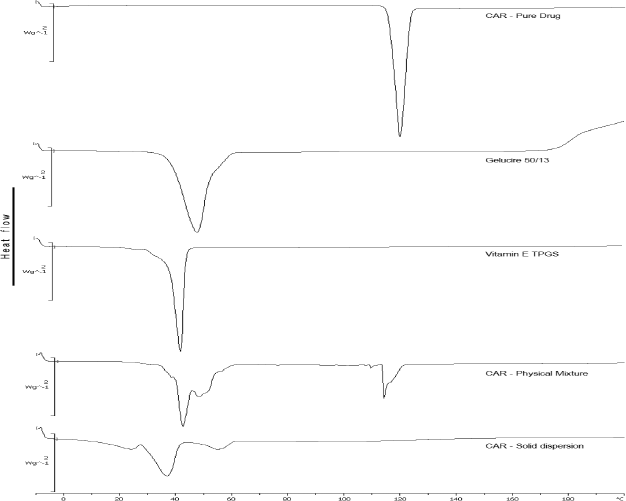 | Figure 1. DSC thermograms of carvedilol, Gelucire 50/13, TPGS, physical mixture, and solid dispersion. [Click here to view] |
In vitro dissolution studies for carvedilol solid dispersions
Phosphate buffer (pH 6.8) was found to be a discriminative dissolution method and was selected for the evaluation of carvedilol solid dispersions. Dissolution profiles for carvedilol and solid dispersion are shown in Figures 2 and 3. Solid dispersions comprised of Gelucire 50/13 show better dissolution profile overall when compared to those containing Gelucire 44/14. The percent of drug release from pure drug and SD4 (1:2 ratio of Gelucire 44/14) after 240 minutes were 16.7% and 36.0%, respectively, indicating a 2.1-fold increase in the solubility and dissolution rate. In comparison, solid dispersions formulated with Gelucire 50/13 were more effective depicting a higher release rate. For example, formulation SD5, presented a release of 22.3% in the first 60 minutes while reaching 41% by the end of 4 hours. The highest amount of carvedilol released was found to be with SD8, where 18.8% of drug was released within the first 60 minutes and then reaching 83% after 4 hours and there is a statistically significant difference in the drug release profiles. Interestingly, SD10, containing a higher ratio of Gelucire 50/13 to drug, exhibited a release of only 72% after 4 hours, notably lower than formulation SD8. A higher concentration of Gelucire 50/13 proved not effective in improving the dissolution rate of carvedilol. It was observed that after coming in contact with media, SD10 formed a dense matrix that proved difficult to dissolve, impeding the rapid dissolution of drug, acting more similar to a controlled release formulation. The in vitro dissolution studies performed clearly suggest that there is a markedly increase in the dissolution rate of carvedilol when Gelucire is incorporated in the formulation, further confirming that solid dispersions enhance the solubility of poorly soluble drugs.
Since Gelucire 50/13 demonstrated better dissolution profiles overall, SD8 was selected to study the synergetic effects in dissolution when incorporating TPGS into the system. To select the proper concentration, solubility studies were first performed by adding different concentrations of TPGS to SD8. As illustrated in Figure 4, the addition of TPGS to carvedilol further enhances the solubility of carvedilol as a function of concentration. To confirm these findings, dissolution studies in phosphate buffer (pH 6.8) were performed for SD8 and SD8+ 10% TPGS (w/w). Side by side comparison of the dissolution profile is shown in Figure 5. After 60 minutes, the percentage dissolved from SD8 was 42% whereas in combination with TPGS was 53%. After 4 hours, SD8+ 10% TPGS dissolution percentage was 88%, significantly higher than the 76% achieved by SD8 further demonstrating that a combination of two surface active excipients can increase the dissolution rate of poorly water-soluble carvedilol. Enhanced dissolution can be a result of different facts including amorphization of drug compound, particle size reduction, and increased wettability and dispersibility.
 | Figure 2. Comparison of dissolution profile of Gelucire 44/14 formulations. [Click here to view] |
 | Figure 3. Comparison of dissolution profile of Gelucire 50/13 formulations. [Click here to view] |
Tablets and capsules were compound with the formulation SD8+ 10% TPGS. Comparison of tablet and capsule dissolution profile are presented in Figure 6. Here, it can be seen that tablet form exhibited a lower dissolution when compared to capsule form. After 4 hours, the maximum concentration achieve by tablet form was 62% which was notably lower than the concentration achieved by capsule form (87%). During the dissolution study of the tablet form, small aggregates were observed in the bottom of the vessel, which may be the reason behind the incomplete dissolution shown. Compression of the powder into a tablet form generated intrinsic high forces that could have melted the semisolid excipients leading to agglomeration of particles affecting the dissolution profile. These aggregates were not observed in the capsule form. This problem might be solved by further formulation development, which unfortunately was out of the scope of this research project.
Stability study
Assay and dissolution results of stability samples are presented in Tables 3 and 4. Initial assay of carvedilol solid dispersion was 97.6%. Percent drug released after 4 hours was 87.5% on day zero. Carvedilol solid dispersion after stored 3 months under accelerated stability conditions presented an assay of 97.6% with a percent drug release of 83.6%. Refrigerated conditions and room temperature storage conditions had shown assay values of 105.6% and 96.6% and a drug release of 84.2% and 85.1%, respectively. Low degradation of carvedilol in the formulation may be due to the inclusion of TPGS that also acts like an antioxidant agent protecting the drug from degradation. The study shows successful results and carvedilol solid dispersion was stable for 3 months under accelerated degradation conditions.
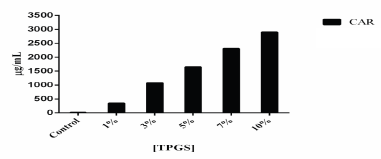 | Figure 4. Solubility studies incorporating different concentrations of TPGS to carvedilol. [Click here to view] |
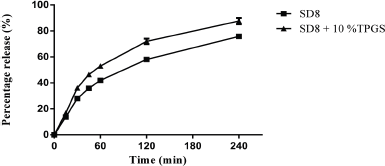 | Figure 5. Comparison of dissolution profile of SD8 and SD8+ 10% TPGS. [Click here to view] |
.png) | Figure 6. Comparison of dissolution profile of tablet and capsule formulations. [Click here to view] |
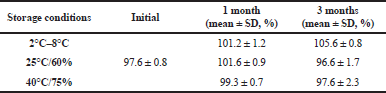 | Table 3. Assay for solid dispersion formulation (SD8) subjected to stability studies. [Click here to view] |
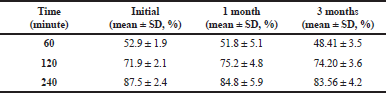 | Table 4. In vitro cumulative percentage drug release for solid dispersion capsules at accelerated stability (40°C/ 75% RH). [Click here to view] |
PAMPA study
PAMPA provides an estimate of the passive transcellular permeability because it is a non-cell based permeability model that lacks transporter and pore mediated permeability. For a drug to travel across the artificial membrane, an adequate lipophilicity (logP) is necessary, similar to that needed to permeate phospholipid membranes by passive diffusion. Effect of Gelucire 50/13 and TPGS on permeability of carvedilol in PAMPA model is shown in Figure 7. The effective permeability (Pe) values for solid dispersion and carvedilol pure drug was found to be 1.29 × 10−7 and 6.13 × 10−8, respectively, which indicate that solid dispersions presented a higher permeability through lipid membranes compared to pure drug. Statistical analysis of permeability coefficient of carvedilol solid dispersion and pure carvedilol powder (p < 0.05) further confirmed the significant increase in the permeability observed for carvedilol when formulated as a solid dispersion with Gelucire 50/13 and TPGS.
Pharmacokinetics study
Extraction of carvedilol from rat plasma by protein precipitation method showed sharp peaks in the chromatogram. The relation between carvedilol concentration and peak area ratios of IS was linear from 10 to 1,000 ng/ml (y = 0.0096x − 0.0357, r2 = 0.9986). Precision and accuracy were calculated based upon the calibration curve (Table 5). The results obtained from the plasma samples after a single freeze-thaw cycle did not indicate any degradation had occurred with a mean percentage concentration of 98.4%, 101.2%, and 98.6% for the standard concentrations of 20, 100, and 500 ng/ml, respectively. Plasma concentration-time profile of Gelucire solid dispersion, Gelucire-TPGS formulation, and the pure drug at a dose of 40 mg/kg carvedilol base are presented in Figure 8, and the corresponding pharmacokinetics parameters are summarized in Table 6. The average maximum peak plasma concentration obtained for Gelucire-TPGS formulation was 581.2 ng/ml, significantly higher than the concentration obtained for carvedilol pure drug, 417.1ng/ml, and Gelucire solid dispersion, 459.8 ng/ml. From peak plasma concentrations, indicating an increase in the extent of absorption. Furthermore, Gelucire solid dispersion had a relatively higher peak plasma concentration than carvedilol pure drug, even if they were not significantly different (459.8 and 417.1 ng/ml, respectively). Increase in the Cmax values from Gelucire-TPGS formulation indicates higher attainable plasma drug concentration with the same dose of the drug. The time to reach maximum plasma concentration (Tmax) for both Gelucire and Gelucire-TPGS formulations was equal to the value obtained for carvedilol pure drug, an indication of fast absorption from the optimized formulation that can be attributed to the rapid dissolution, which was in accord with the results of in vitro dissolution. This will lead to a faster onset of action. Area under the curve (AUC) values for both solid dispersions was markedly higher when compared to pure drug. However, Kel and t1/2 values of both solid dispersions were not significantly different from pure drug. The pharmacokinetic parameters indicate that the relative bioavailability, ratio of the exposure from one formulation relative to another different formulation, was 169% for Gelucire-TPGS formulation and 112% for Gelucire solid dispersion when compared to pure carvedilol.
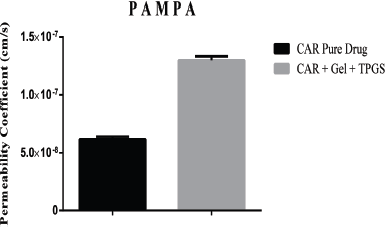 | Figure 7. Comparative PAMPA permeability: effect of solid dispersion on permeation. [Click here to view] |
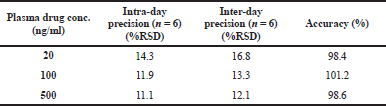 | Table 5. Bioanalytical method validation results for determination of carvedilol in rat plasma. [Click here to view] |
From the pharmacokinetic analysis, it can be concluded that the in vivo studies mimic the in vitro results. In vitro results demonstrated considerable difference in the percentage drug release between pure drug and solid dispersion formulation, similar differences were observed in Cmax, Tmax, and AUC between pure drug and solid dispersion formulations (Fig. 8). Solid dispersions systems provided a great improvement for in vivo absorption of carvedilol, supporting other studies that used solid dispersions to enhance the bioavailability of poorly water soluble drugs. Inclusion in the formulation of a surface active agent such as TPGS, further enhances oral bioavailability of carvedilol.
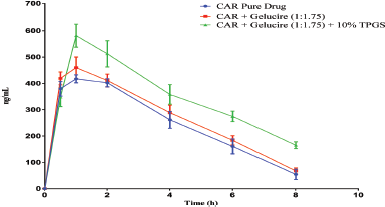 | Figure 8. Mean plasma concentration-time profile of carvedilol after single oral administration of pure drug. [Click here to view] |
.png) | Table 6. Pharmacokinetic parameters of carvedilol formulations after oral administration to male Sprague-Dawley rats. [Click here to view] |
DISCUSSION
Carvedilol exhibits poor bioavailability because of its poor solubility and extensive first-pass metabolism. Solid dispersions are systems in which a hydrophobic drug is dispersed in a hydrophilic matrix in order to improve dissolution properties and bioavailability (Mohan and Gundamaraju, 2015). Furthermore, the incorporation of surface active agents into solid dispersions can improve the solubility of class II drugs by its improving wettability. In the present study, solid dispersions using Gelucire and TPGS were formulated to investigate its potential in increasing carvedilol solubility and bioavailability.
Carvedilol solid dispersions were prepared using Gelucire 44/14 and Gelucire 50/13 as hydrophilic matrix. TPGS, a surface active agent derived from vitamin E, was incorporated to further enhance the dissolution profile and as a potential inhibitor of P-gp to enhance oral bioavailability of carvedilol. To the resultant solid dispersions MCC and amorphous fumed silica were added to obtain a free flowing powder that was filled into size 1 capsules. Solid dispersion formulations were tested for in vitro dissolution and compared against pure carvedilol. Gelucire 50/13 in a ratio of 1 to 1.75 (drug to Gelucire) showed a drug release of more than 80% in 4 hours, while the drug release from pure carvedilol was less than 20% during the same time. Hence, this formulation was selected for further studies.
The solubility of carvedilol with different concentrations of TPGS was also investigated. Addition of TPGS resulted in increased solubility in a direct proportion. As a result of these studies, 10% of TPGS was incorporated to Gelucire 50/13 formulation to further enhance the dissolution of carvedilol. In vitro dissolution studies resulted in a significantly higher release of carvedilol from the formulation containing TPGS (88%). DSC analysis revealed that there was a change in the physical nature of carvedilol when formulated as a solid dispersion, an indication that it might be present in an amorphous state. In vitro permeability studies using PAMPA model revealed that Gelucire-TPGS solid dispersion showed a higher permeability coefficient compared to pure drug. The enhancement of in vitro permeation appears to be due to combined effect of improved solubility/dissolution and increased passive diffusion.
Stability studies of solid dispersion capsules were performed at different storage conditions for a period of 3 months. There were no significant differences in the assay and dissolution data at 120 and 240 minutes period after 3 months. In vivo evaluation of Gelucire, Gelucire-TPGS and pure drug (carvedilol) were studied using male Sprague-Dawley rats. Pharmacokinetic studies in rats revealed that plasma average peak concentration of carvedilol after administration of Gelucire-TPGS formulation resulted in higher peak concentrations compared to Gelucire formulation and pure drug, 581, 459, and 417 ng/ml, respectively. The overall exposure (AUC) of Gelucire-TPGS formulation was significantly higher than that of carvedilol pure drug. The relative bioavailability of carvedilol was 169% higher compared to pure drug suspension. The results suggest that the enhanced oral bioavailability of carvedilol in the Gelucire-TPGS solid dispersion was due to the marked increase in the absorption of the drug through its improved solubility and dissolution. Hence, we conclude that Gelucire 50/13 solid dispersion with the incorporation of TPGS resulted in increased bioavailability of poorly soluble drug carvedilol.
CONCLUSION
Within the present investigation, a free flowing powder solid dispersion incorporating Gelucire 50/13 and TPGS for oral administration of carvedilol was developed. The optimized formulation presented rapid dissolution rate over the course of 4 hours. Additionally, passive permeability studies in PAMPA model depicted improved permeability of the formulation when compared to free drug. The optimized formulation was found to be stable at accelerated conditions up to 3 months. Increased bioavailability of carvedilol-Gelucire-TPGS was shown with high levels of carvedilol found in plasma, representing a 1.4-fold increase in AUC. Solid dispersions are a suitable technology to improve wettability and increase oral bioavailability of poorly soluble drugs. This investigational study results further confirm that third generation solid dispersions incorporating two different nonionic surfactants represent a promising platform technology for improving the oral bioavailability of carvedilol.
ACKNOWLEDGMENT
The authors express their sincere gratitude to Western University of Health Sciences, Pomona, California for providing the funding and facilities.
FINANCIAL SUPPORT
None.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification—the correlation of in-vitro drug product dissolution and in-vivo bioavailability. Pharm Res, 1995; 12(3):413–20. CrossRef
Bart J, Dijkers ECF, Wegman TD, de Vries EGE, van der Graaf WTA, Groen HJM, Vaalburg W, Willemsen ATM, Hendrikse NH. New positron emission tomography tracer [11C]carvedilol reveals P-glycoprotein modulation kinetics, Br J Pharmacol, 2005; 145(8):1045–51. CrossRef
Chen X, Murawski A, Patel K, Crespi C, Balimane P. A novel design of artificial membrane for improving the PAMPA Model. Pharm Res, 2008; 25(7):1511–20. CrossRef
Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev, 2008; 60(6):717–33. CrossRef
Dunn C, Lea A, Wagstaff A, Carvedilol. Drugs, 1997; 54(1):161–85. CrossRef
Huang Y, Dai WG. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B, 2014; 4(1):18–25. CrossRef
Kovvasu SP, Kunamaneni P, Kunderu RS. Cyclodextrins and their application in enhancing the solubility, dissolution rate and bioavailability. Innoriginal Int J Sci, 2018a; 5(5):25–34.
Kovvasu SP, Chowdary KPR. Formulation development and in vivo evaluation of pioglitazone inclusion complexes: a factorial study. Int J App Pharm, 2018b; 10(3):49–55.
Lee W, Woo ER, Choi JS. Effects of myricetin on the bioavailability of carvedilol in rats. Pharm Biol, 2012; 50(4):516–22. CrossRef
Mohan A, Gundamaraju R. In vitro and in vivo evaluation of fast-dissolving tablets containing solid dispersion of lamotrigine. Int J Pharmaceutical Invest, 2015; 5(1):57–64. CrossRef
Pokharkar VB, Mandpe LP, Padamwar MN, Ambike AA, Mahadik KR, Paradkar A. Development, characterization and stabilization of amorphous form of a low Tg drug. Powder Tech, 2006; 167(1):20–5. CrossRef
Savjani KT, Gajjar AK, Savjani JK. Drug Solubility: importance and enhancement techniques. ISRN Pharmaceutics, 2012; 1–10. CrossRef
Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharmaceutical Sci, 1999; 88(10):1058–66. CrossRef
Tian B, Wang X, Zhang Y, Zhang K, Zhang Y, Tang X. Theoretical prediction of a phase diagram for solid dispersions. Pharm Res, 2015; 32(3):840–51. CrossRef
Yasir M, Asif M, Kumar A, Aggarval A. Biopharmaceutical classification system: an account. Int J PharmTech Res, 2010; 2(3):1681–90.
Zhang M, Li H, Lang B, O’Donnell K, Zhang H, Wang Z, Dong Y, Wu C, Williams IRO. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur J Pharm Biopharm, 2012a; 82(3):534–44. CrossRef
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials, 2012b; 33:4889–906. CrossRef