
INTRODUCTION
One-third of the global population has been latently infected with TB (LoBue et al., 2009). The WHO reported that an estimated 10 million people fell ill with TB worldwide, and India accounted for 27% of the global TB cases in the year 2018 (World Health Organization, 2019). TB is predominantly an infectious disease caused by a group of mycobacterial species called Mycobacterium tuberculosis complex. It is primarily a pulmonary pathogen but can infect other parts of the human body (Jassal and Bishai, 2009; Kumar et al., 2017c). The incidence of drug resistance against key anti-TB drugs is the leading challenge for the global TB control (Centers for Disease Control and Prevention, 2006; Gandhi et al., 2006; Prasad, 2010; Wright et al., 2009; Zignol et al., 2016). The combination therapy of anti-TB drugs and directly observed treatment short-course was an excellent initiative to prevent the emergence of drug-resistant TB (Johnson, 2007). However, drug-resistant TB has emerged and its prevalence is increasing day by day. Improper, incomplete, or interrupted treatment courses with anti-TB drugs are the major reasons behind the acquired drug resistance (World Health Organization, 2008; Borrell and Gagneux, 2009; World Health Organization, 2014). This situation leads to MDR-TB, where the bacilli resist both rifampicin and isoniazid, which has been virtually reported in all settings. The second-line anti-TB drug treatments for MDR-TB cases are comparatively less effective with more side effects (Falzon et al., 2017). In 2015, the WHO estimated that there were 580,000 new cases of MDR-TB and that 250,000 MDR-TB deaths occurred globally. The estimated global MDR/RR TB burden was 4.1% for new cases and 19% for previously treated cases for the year 2016 (World Health Organization, 2017). Among there MDR, 8.5% extensively drug resistant TB (bacilli having additional resistance with fluoroquinolones and one of the second line injectable anti-TB drugs) and 22% fluoroquinolones resistant cases have been reported in the global TB report 2018 (World Health Organization, 2018). Human immunodeficiency virus coinfection with TB made the situation comparatively more hazardous than active TB infection alone (Gagneux, 2012).
Mycobacteria have MEP pathways that are involved in the production of a group of compounds called isoprenoids. The group consists of more than 40,000 compounds that play a crucial role in the growth and development of living organisms (Li and Wang, 2016). These compounds function as both primary and secondary metabolites (Frank and Groll, 2016; Sacchettini and Poulter, 1997). These isoprenoids are significantly involved in cellular metabolism, such as electron transport system, cell respiration, photosynthesis, membrane biosynthesis, cell signaling, and so on (Bouvier et al., 2005; Heuston et al., 2012). MTB has essential isoprenoids like menaquinone (which participates in oxidative phosphorylation) and polyprenyl phosphate (which synthesizes the components of the cell wall like arabinogalactan and lipoarabinomannan) (Bouvier et al., 2005; Brennan, 2003). These types of isoprenoids are synthesized by polymerization of five carbon isoprene units of IPP and its isomer dimethylallyl pyrophosphate (Rodrıguez-Concepción and Boronat, 2002). The known biosynthesis pathways for isoprenoids in humans and bacteria are different. In humans, it follows the mevalonate pathway, while in bacteria they follow the MEP pathway. Additionally, it is interesting that the enzymes involved in the MEP pathway have no homology with the proteins of humans (Sassetti et al., 2003). Several enzymes are involved in the MEP pathway; however, IPP is observed as one of the essential enzymes for the survival for the mycobacterium (Eoh et al., 2007; Eoh et al., 2009). This detail prompts us to consider the IspD enzyme as an ideal target for inhibiting the mycobacterial MEP pathway and to develop antimycobacterial molecules (Eoh et al., 2009). With regard to MDR-TB, there is a need for a novel drug discovery with a new approach which can be used in the combination therapy to shorten the duration of treatment (Andries et al., 2005; World Health Organization, 2014).
In this study, we have evaluated and analyzed the specific interactions of the IspD enzyme with the available ligands by using the in silico molecular docking tool. Despite having fosmidomycin and aryl bis-sulfonamide as efficient inhibitors against IspD (Zhang et al., 2011) and IspF (Thelemann et al., 2015), other molecules are also being considered in this study to observe their possible interactions with MTB IspD.
METHODS AND MATERIALS
PDB file retrieval
The PDB structure of the M. tuberculosis IspD enzyme was complex with positive control (CTP) and Mg2+ ion. The 3Q7U.pdb was retrieved from the research collaboratory for structural bioinformatics website (Berman et al., 2000). The mycobacterial IspD is a homodimer, wherein each subunit consists of 231 amino acid residues. Each subunit contains two crucial domains, which includes the larger globular domain and the smaller β-domain. Here, the chain A of 3Q7U.pdb file was selected for the computational study. The β-domains of the monomers are mainly responsible for the formation of the dimer. The attached CTP and Mg2+ ion were deleted from the structure, followed by refinement of the protein subunit.
Protein simulation
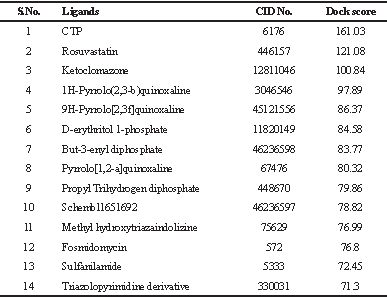
The catalytic pocket, which actively participates in interaction with ligands, mainly consists of polar amino acid residues. The 3-D structure was further refined by using the CHARMM inbuilt in DSv3.5, which minimizes the energy of the macromolecule (Brooks et al., 1983; Kumar et al., 2017a; Visualizer, 2012). A total of 13 ligands along with one CTP were used for this study. The 3-D structures of these ligands were taken from PubChem in the SDF file (Kim et al., 2016; Rana et al., 2017). The details of the ligands with their PubChem CID codes are listed in Table 1. Further simulation of the protein was carried out by using the SHAKE algorithm. The simulation process was carried out in 2000 steps of the steepest descent minimization techniques at 300K (Kräutler et al., 2001; Kumar et al., 2017b).
Conserved domain identification and binding site
Each protein has its conserved domain in which the ligands show their chemical activities. All the ligands were optimized before docking by using the prepare ligand tool DSv3.5. The ligand-binding site module is an authentic program that can predict and can suitably characterize the binding site module with all its functional residues in the protein by using the DSv3.5. The entire amino acid sequence of 3Q7U_A was selected and with the help of the CHARMM force field, and the protein coordinate was prepared. The binding site for the natural substrate was selected for docking (Kant et al., 2018).
Docking process
Molecular docking was carried out on the prepared protein and stimulated ligands using the DSv3.5 computational package. Docking occurred between the prepared protein and ligands at the defined site (coordinates: X-26.622, Y-4.23, and Z-14.34) by using the LibDock algorithm. The selected ligands were processed and optimized before docking. The protein interacted with each optimized compound in several poses, and their interactions are evaluated in terms of the LibDock score. The highest score for a specific ligand projects the best suited pose. The LibDock possesses specific physicochemical properties of the ligands that direct docking according to the equivalent features present in the protein binding sites. The docking analysis discovered that rosuvastatin (RST) is the best scoring molecule that can effectively inhibit the domain of the IspD enzyme (Kumar et al., 2018a; Kumar et al., 2019; Kumari et al., 2019; Mahato et al., 2017).
RESULTS AND DISCUSSION
Genetic mutation is assumed to be the reason behind most of the drug resistance cases. For example, rifampicin targets the β-subunit of the RNA polymerase and eventually blocks transcription. The rpoB gene identifies the β-subunit of the RNA polymerase. The specific genetic mutation in the sequence of the rpoB gene can make the bacilli resistant to rifampicin. Additionally, antibiotic members of the same group of drugs having the same target can show a certain level of cross-resistance. Therefore, it is more important to find new targets for drug development against TB. The present research study targeted another enzyme, i.e., IspD, which is essential for bacterial survival.
 | Table 1. List of ligands used for molecular docking and their highest dock score with IspD. [Click here to view] |
.png) | Figure 1. Three dimensional model of the IspD protein produced by the DSv3.5. α helices are in red, β sheets are in blue, and FL are in grey. [Click here to view] |
Modeled protein prediction and assessment using homology modeling approaches
Chain A of the 3-D structure of 3Q7U has been used in this study. The monomer chain A has one active site in which the optimized ligands were docked for their optimum activities. The structure has seven alpha helixes (α), three small 310 helixes (η), ten beta sheets (β), and three flexible loops (FL) (Fig. 1). The sequence of amino acids residues is shown according to Kabsch and Sander’s method in Figure 2. The 3D–1D score assessed by Verify3D described 85.84% of the amino acid residues, with an average 3D–1D score of ≥ 0.2. Notably, the overall model excellence was illustrated by the ERRAT plot analysis, which showed that nearly 91.705% of the residues were found below the 95% error cutoff limits (Fig. 3). The 2-D protein structure showed interactions between Gly16, Arg83, Thr84, and Tyr190 with RST (Fig. 4). It also showed weak interactions that probably govern the binding of the RST to the catalytic pocket in IspD. The receptor which binds with ligand formed six intermolecular hydrogen bonds with electrostatic interaction and Van der Waal’s force, which enabled suitable interaction between the protein and the ligand.
Protein–ligand interaction
The in-depth in silico analysis revealed the prepared model suitably interacted with the proposed ligands. The protein–ligand interaction was assessed and validated by the DSv3.5. The ligands and protein subunit were optimized, and molecular docking was carried out at the natural active site of the protein. A range of dock scores were observed for each ligand with different poses. The focus was on the ligand with the highest dock score after positive control. We observed that RST had the highest LibDock score of 121.08. RST is a member of the statin family that is used to control the formation of cholesterol in humans. It competitively inhibits the HMG Co-A reductase, a critical enzyme of the mevalonate pathway for isoprenoids production (Prinz et al., 2008). Details of all the studied ligands with their maximum LibDock score are shown in Table 1.
The modeled IspD protein strongly interacted with the RST ligand. The crucial active site residues Gly16, Arg83, Thr84, and Thy190 played a vital role in the protein–ligand stabilization process (Fig. 5). The amino acid residue Gly16 was present in the FL1, and Arg83 and Thr84 were found within the α3-helix. The surface view of the RST–IspD interactions is shown in Figure 6. Ketoclomazone and 1H-pyrrolo-(2,3-b)-quinoxaline are the other essential compounds with an LibDock score of 100.84 and 97.89, respectively, followed the RST–IspD interaction. The atomic interaction between the RST and IspD is presented in Table 2.
Obiol-Pardo et al. (2010) modeled the IspD of M. tuberculosis to derive a reliable model for use in the structure-based drug design. Varikoti et al. (2012) attempted to structure the de novo design of possible inhibitor molecules for docking at the active site of the IspD enzyme. The docking score observed for RST in this study was slightly lesser than that observed for the natural substrate. However, it does not reflect the affinity of RST to be less than CTP for this protein. We used the IspD structure that was already bound to the CTP and then removed the CTP. Therefore, there are conformational changes in the IspD structure that is bound and unbound to CTP. Recently, the growth retarding effect of RST has been observed on the MTB culture (Kumar et al., 2018b).
.png) | Figure 2. Amino acid sequence of 3Q7U. [Click here to view] |
.png) | Figure 3. Overall quality checked by ERRAT. [Click here to view] |
CONCLUSION
The current work suggests that RST is a potent molecule for binding the enzyme IspD to other vital molecular interactions at its active site. However, at present, new drugs for TB treatment are urgently required due to the rapid emergence of drug resistance against available anti-TB drugs and identification of RST, or its structural analog, as a novel anti-TB agent, can play a key role in eliminating TB worldwide. However, the study on the binding RST to other sites of the enzyme and further in vitro evaluations will be the future prospects of research.
.png) | Figure 4. The two-dimensional IspD–RST interaction. [Click here to view] |
.png) | Figure 5. Protein–ligand interaction of IspD–RST showing intermolecular H-bonding. [Click here to view] |
.png) | Figure 6. Interactions of IspD and RST (surface view). [Click here to view] |
.png) | Table 2. Estimated intermolecular H-bonding between RST and IspD. [Click here to view] |
ACKNOWLEDGMENTS
This research work was supported by the Biomedical Informatics Center RMRIMS, Agamkuan, Patna. I thank the research team of the Department of Biotechnology, College of Commerce, Arts and Science, Patna, for their motivation and support.
ABBREVIATIONS
CID: Compound identifier; DSv3.5: Discovery studio version 3.5; IPP: Isopentenyl pyrophosphate; IspD: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; MDR-TB: Multidrug-resistant TB; MEP: Methylerythritol phosphate; MTB: Mycobacterium tuberculosis; RNA: Ribonucleic acid; RST: Rosuvastatin; TB: Tuberculosis; WHO: World Health Organization. PDB: Protein Data Bank; CHARMM: Chemistry at Harvard Macromolecular Mechanics; SDF: Spatial Data File; HMG: 3-hydroxy-3-methyl-glutaryl.
CONFLICT OF INTEREST
None.
FUNDING
None.
REFERENCES
Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler, H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science, 2005; 307(5707):223–7. CrossRef
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shinyalov IN, Bourne PE: the protein data bank. Nucleic Acids Res, 2000; 28(1):235–42. CrossRef
Borrell S, Gagneux S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis [State of the art]. Int J Tuberc Lung Dis, 2009; 13(12):1456–66.
Bouvier F, Rahier A, Camara B. Biogenesis, molecular regulation and function of plant isoprenoids. Prog Lipid Res, 2005; 44(6):357–429. CrossRef
Brennan PJ. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis, 2003; 83(1):91–7. CrossRef
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem, 1983; 4(2):187–217. CrossRef
Centers for Disease Control and Prevention. Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs–worldwide, 2000-2004. MMWR Morb Mortal wkly Rep, 2006; 55(11):301.
Eoh H, Brennan PJ, Crick DC. The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis, 2009; 89(1):1–11. CrossRef
Eoh H, Brown, AC, Buetow L, Hunter WN, Parish T, Kaur D, Brennan PJ, Crick DC. Characterization of the Mycobacterium tuberculosis 4-diphosphocytidyl-2-C-methyl-D-erythritol synthase: potential for drug development. J Bacteriol, 2007; 189(24):8922–7. CrossRef
Falzon D, Schunemann HJ, Harausz E, Gonzalez-Angulo L, Lienhardt C, Weyer K. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur Respir J, 2017; 49(3):1602308. CrossRef
Frank A, Groll M. The methylerythritol phosphate pathway to isoprenoids. Chem Rev, 2016; 117(8):5675–703. CrossRef
Gagneux S. Host–pathogen coevolution in human tuberculosis. Phil Trans R Soc B, 2012; 367(1590):850–9. CrossRef
Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet, 2006; 368(9547):1575–80. CrossRef
Heuston S, Begley M, Gahan CG, Hill C. Isoprenoid biosynthesis in bacterial pathogens. Microbiology, 2012; 158(6):1389–1401. CrossRef
Jassal M, Bishai WR. Extensively drug-resistant tuberculosis. Lancet Infect Dis, 2009; 9(1):19–30. CrossRef
Johnson R. “Understanding the mechanisms of drug resistance in enhancing rapid molecular detection of drug resistance in Mycobacterium tuberculosis.” PhD diss., Stellenbosch: University of Stellenbosch, 2007.
Kant V, Sahoo G, Kumar M, Dikhit M, Sinha S, Das P. Designing of peptide and non-peptide inhibitors against leishmania cysteine-synthase (Ld-OASS). Int J Infect Dis, 2018; 73:301. CrossRef
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang, J, Yu B, Zhang J, Bryant SH. PubChem substance and compound databases. Nucleic Acids Res, 2016; 44(D1):D1202–13. CrossRef
Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem, 2001; 22(5):501–8. CrossRef
Kumar M, Rana S, Kumar H, Kumar J, Mansuri R, Chandra Sahoo G. Inhibition of product template (PT) domain of aflatoxin producing polyketide synthase enzyme of Aspergillus parasiticus. Lett Drug Des Discov, 2017a; 14(7): 811–8. CrossRef
Kumar M, Rana S, Kumar H, Kumar P, Dikhit MR, Mansuri R, Kumar J, Sahoo GC. Computational, structural and functional aspects of hypothetical protein of Aspergillus flavus Pheromone Receptor Pre-A (PRP-A). J Appl Pharm Sci, 2017b; 7(07):089–97.
Kumar M, Sahoo G, Topno R, Pandey K, Ravidas V, Dikhit M, Kumar H, Das P. Molecular interaction of antifungal compounds with fungal type I Polyketide synthase and preventing hepatocarcinogenesis. Int J Infect Dis, 2018a; 73:285. CrossRef
Kumar M, Topno RK, Dikhit MR, Sahoo GC, Madhukar M, Pandey K, Das P. Molecular docking studies of chloroquine and its derivatives against P23pro-zbd domain of chikungunya virus: Implication in designing of novel therapeutic strategies. J Cell Biochem, 2019; 120(10):18298–308. CrossRef
Kumar P, Goswami P, Kumari R, Kumar M, Kumar S. Comparative Study of LED fluorescent microscopy with the conventional ZN based microscopy for diagnosis of microbiologically confirmed TB cases. Int J Adv Res, 2017c; 5(6):1083–6. CrossRef
Kumar P, Kumar M, Kumar H, Kumar J, In vitro effects of rosuvastatin on Mycobacterium tuberculosis. Int J Curr Res Life Sci, 2018b; 7(4):1885–7.
Kumari A, Kumar M, Sahoo GC, Kumar J. In silico prediction of Glabridin potency against Human Tyrosinase in hyperpigmentation condition. Int J Res Anal Rev, 2019; 6(2):332–41.
Li Y, Wang G. Strategies of isoprenoids production in engineered bacteria. J Appl Microbiol, 2016; 121(4):932–40. CrossRef
LoBue P, Sizemore C, Castro KG. Plan to combat extensively drug-resistant tuberculosis: recommendations of the Federal Tuberculosis Task Force. MMWR Recomm Rep, 2009; 58(RR-3):1–43.
Mahato JP, Rana S, Kumar M, Sarsaiya S. Homology modeling of Leishmania donovani enolase and its molecular interaction with novel inhibitors. J Pharm Bioallied Sci, 2017; 9(2):99.
Obiol-Pardo C, Cordero A, RubioMartinez J, Imperial S. Homology modeling of Mycobacterium tuberculosis 2-C-methyl-D-erythritol-4-phosphate cytidylyltransferase, the third enzyme in the MEP pathway for isoprenoid biosynthesis. J Mol Model, 2010; 16(6):106173. doi:10.1007/s008940090615x. CrossRef
Prasad R. Multidrug and extensively drug-resistant TB (M/XDR-TB): problems and solutions. Indian J Tuberc, 2010; 57(4):180–91.
Prinz V, Laufs U, Gertz, K, Kronenberg G, Balkaya M, Leithner C, Lindauer U, Endres M. Intravenous rosuvastatin for acute stroke treatment: an animal study. Stroke, 2008; 39(2):433–8. CrossRef
Rana S, Mahat JP, Kumar M, Sarsaiya S. Modeling and docking of cysteine protease-A (CPA) of leishmania donovani. J Appl Pharm Sci, 2017; 7:179–84.
Rodrıguez-Concepción M, Boronat A. Elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis in bacteria and plastids. A metabolic milestone achieved through genomics. Plant Physiol, 2002; 130(3):1079–89. CrossRef
Sacchettini JC, Poulter CD. Creating isoprenoid diversity. Science, 1997; 277(5333):1788–9. CrossRef
Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol, 2003; 48(1):77–84. CrossRef
Thelemann J, Illarionov B, barylyuk K, Geist, J, Kirchmair J, Schneider P, Anthore L, Root K, Trapp N, Bacher A, Witschel M. Aryl Bis-sulfonamide inhibitors of IspF from arabidopsis thaliana and plasmodium falciparum. ChemMedChem, 2015; 10(12):2090–8. CrossRef
Varikoti RA, Gangwal R, Dhoke G, Ramaswamy VK, Sangamwar A. Structure based de novo design of IspD inhibitors as anti-tubercular agents. Nature Precedings. 2012; 2:1–1. CrossRef
Visualizer DS. Release 3.5. Accelrys Inc, San Diego, CA, 2012.
Weyer K, Dennis Falzon D, Jaramillo E, Zignol M, Mirzayev F, Raviglione M. Drug-resistant tuberculosis: what is the situation, what are the needs to roll it back. AMR Control, 2017; 20:60–7.
World Health Organization, global TB report 2017.
World Health Organization, global TB report 2018.
World Health Organization, global TB report 2019.
World Health Organization. Companion handbook to the WHO guidelines for the programmatic management of drug-resistant tuberculosis, 2014.
World Health Organization. Policy guidance on drug-susceptibility testing (DST) of second-line antituberculosis drugs. World Health Organization, Geneva, Switzerland, 2008.
Wright A, Zignol M, Van DA, Falzon D, Gerdes SR, Feldman K, Hoffner S, Drobniewski F, Barrera L, Van Soolingen D, Boulabhal F, Paramasivan CN, Kam KM, Mitarai S, Nunn P, Raviglione M. Epidemiology of antituberculosis drug resistance 2002–07: an updated analysis of the Global Project on Anti-Tuberculosis Drug Resistance Surveillance. Lancet, 2009; 373(9678):1861–73. CrossRef
Zhang B, Watts KM, Hodge D, Kemp LM, Hunstad DA, Hicks LM, Odom AO. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry, 2011; 50(17):3570–7. CrossRef
Zignol M, Anna S, Falzon D, Gemert W-V, Wright A, Deun A-V, Portaels F, Laszlo A, Espinal MA, Mendez AP, Bloom A, Aziz MA, Weyer K, Jaramillo E, Nunn P, Floyd K, Raviglione MC. Twenty years of global surveillance of antituberculosis-drug resistance. N Engl J Med, 2016; 375(11):1081–9. CrossRef