
INTRODUCTION
The process of the development of arterioles from pre-existing vessels is known as angiogenesis, which plays a climacteric role in proliferation, migration, and survival of endothelial cells (Carmeliet and Jain, 2011). Stimulation of angiogenesis is amidst the hallmarks of tumor growth and cancer (Fouad and Aanei, 2017; Zhao and Adjei, 2015). Vascular endothelial growth factors (VEGFs) and vascular endothelial growth factor receptors (VEGFRs) have a remarkable impact on angiogenesis. Five VEGFs, namely VEGFA, VEGFB, VEGFC, VEGFD, and placental growth factor (PLGF), are involved in the activation of VEGFRs signaling cascade. VEGFRs consist of three receptors, namely VEGFR-1 (Flt-1), VEGFR-2 [Flk-1/kinase domain receptor (KDR)], and VEGFR-3 (Flt-4) (Takahashi and Shibuya, 2005). Among the VEGFR family, KDR is a well-established receptor for the discovery of novel antineoplastic agents (Modi and Kulkarni, 2019). Additionally, the disproportional elevation of VEGFs has been observed due to the activation of oncogenes, loss of tumor suppressor function, and alterations in glucose or oxygen levels. In contrast, autophosphorylation of VEGFR-2 in cancer has been observed due to the overexpression of VEGFs. VEGFR-2 signaling cascade and its role in cancer are shown in Figure 1. Furthermore, several small molecule VEGFR-2 modulators have been developed successfully, some of which are in the clinical trials (Fig. 2) (Frampton, 2012; Harris et al., 2008; Ho and Jonasch, 2011; Roskoski, 2007; Woo and Heo, 2012; Yakes et al., 2011). However, the failure of VEGFR-2 inhibitors in the clinic can be due to both acquired and intrinsic resistance. The occurrence of resistance is due to redundant signaling of receptors, development of hypoxia tolerant tumor cells, hypoxia-resistant malignant clones’ selection, elevation in circulating nontumor proangiogenic factors, and mutations of endothelial cells (Abdullah and Perez-Soler, 2012; Ellis and Hicklin, 2008; Jayson et al., 2016). Furthermore, toxicity associated with VEGFR-2 inhibitors includes thromboembolic complications, proteinuria, hemorrhage, anal fistula, gastrointestinal (GI) perforation, posterior reversible encephalopathy syndrome, hand–foot skin reaction, oral mucositis, diarrhea, thyroid disease, and bone marrow suppression (Kumar et al., 2009; Shepard and Garcia, 2009). These indicate that the discovery of selective VEGFR-2 inhibitors that can overcome the problem of resistance and toxicity remains a considerable task.
 | Figure 1. VEGFR-2 signaling cascades and its role in cancer. FAK = Focal Adhesion Kinase; HPC = Hematopoietic Progenitor Cell; DAG = Diacylglycerol; HSP-27 = Heat-Shock Protein-27; eNOS = Endothelial Nitric Oxide Synthase; MAPK = Mitogen-Activated Protein Kinase; PI3K = Phosphatidylinositol 3′ Kinase; PKC = Protein Kinase C; TSAd = T-cell-specific Adaptor molecule. PLCγ, Phospholipase C-γ; SHB, SH2, and β-cells. *Figure prepared using online tool BioRender (https://biorender.com). [Click here to view] |
3D-QSAR and pharmacophore modeling is a type of ligand-based drug design approach and a widely used method for designing novel analogues with potent inhibitory activity against the biological target. 3D-QSAR enables the identification of pharmacophoric descriptors like hydrophobic, steric, electrostatic, H-bond donor, and acceptor, responsible for the interaction of the molecules with the active site of the receptor. In comparative molecular field analysis (CoMFA), steric and electrostatic descriptors (independent variables) are correlated with the inhibitory activity of ligands (Cramer et al., 1988; Zhu et al., 2005). In 1994, comparative molecular similarity indices analysis (CoMSIA) method was instigated by Klebe et al. (1994) It is an amended technique of CoMFA in which H-bond donor and acceptor, hydrophobic, electrostatic and steric descriptors are correlated with the inhibitory activity of ligands. The most prominent obstacle during drug discovery is the insufficient ADME (Absorption, Distribution, Metabolism, Excretion) parameters of drug molecules. Therefore, a computer-aided drug design approach with the prediction of ADME can rectify the problem of developing new hits during the drug discovery process.
 | Figure 2. Structures of Food and Drug Administration (FDA)-approved VEGFR-2 inhibitors. [Click here to view] |
Naphthyridine and pyridopyrimidine analogous are well-known heterocyclic compounds for cancer chemotherapy. Wang et al. (2020) reported pyridine derivatives bearing 1,6-naphthyridine scaffold as MET and VEGFR-2 inhibitors using scaffold hopping approach (MET, IC50 = 9.8; VEGFR-2 IC50 = 8.8 nM). Kiselev et al. (2010) reported Dibenzo[c,h][1,6]naphthyridines as anticancer agents targeting topoisomerase I enzyme. Salerno et al. (2019) reported Pyrido [3′, 2′: 5, 6] thiopyrano [4, 3-d]pyrimidine analogues as multitargeted tyrosine kinase inhibitors with potent VEGFR-2 inhibitory activity, IC50 = 16 nM. The US20160096832 invention pertains to the synthesis of 7,8-dihydropyrido[4,3-d]pyrimidine derivatives as TKIs for the treatment of cancer (Allergan, Inc., US20160096832, 2016). The US20160096837 invention discloses the synthesis of pyrazolo[1,5-a]pyrido[3,4-e]pyrimidine analogues as tyrosine kinase inhibitors (TKIs) (Allergan, Inc., US20160096837, 2016). With this fact in the current research, CoMFA and CoMSIA models were established for a series of 1,6-naphthyridine and pyridopyrimidine analogues to specify the regions surrounding the molecule where modification might be possible to enhance their activity against VEGFR-2. The developed models were validated using the external validation method and were used further to study contour maps and the structure–activity relationship (SAR). Lastly, the CoMSIA model was used for designing novel analogues and predictions of their inhibitory activity.
MATERIAL AND METHODS
Preparation of data set, molecular modeling, and alignment
A reported series of 1,6-naphthyridine and pyridopyrimidine analogues as VEGFR-2 inhibitors was taken for the 3D-QSAR study (Thompson et al., 2005). Chemical structures of 1,6-naphthyridine and pyridopyrimidines analogues and their IC50 values against VEGFR-2 are given in Table 1. The 1,6-naphthyridine and pyridopyrimidines analogues were alienated into a training set (27 analogues; ~70%) and a test set (14 analogues; ~30%). Analogues of the test set contain full ranged biological activities like the training set. Analogues of training and test sets have been utilized to develop and validate the 3D-QSAR models. IC50 values of all the analogues were transformed into pIC50 (–logIC50) values; the development of CoMFA and CoMSIA models was carried out using these transformed pIC50 values. SYBYL-X 1.2 (Tripos Inc, St. Louis, MO) software was used to carry out the molecular modeling study. Three-dimensional (3D) structures of 1,6-naphthyridine and pyridopyrimidine analogues were constructed in the Sketch module of SYBYL X Molecular Modeling Software (2012), charged using Gasteiger Huckel, and energy was minimized using the Tripos molecular mechanics force field (Basu et al., 2009). Distilled alignment among the series was done by sorting the most potent analogue (20) as a template structure (Fig. 3 and 4).
CoMFA and CoMSIA field generation
For the calculation of CoMFA and CoMSIA fields, a 3D cubic lattice (defined with a grid spacing of 2 Å extended to 4 Å units) beyond the aligned molecules in X, Y, Z directions was used. At the lattice intersection, electrostatic and steric interaction descriptors were evaluated by Lennard-Jones and Columbic potentials. In CoMFA, electrostatic and steric descriptors were derived using Sp3 carbon atom (van der Waals radius of 1.52 Å and +1.0 charge) as a probe atom. However, the cutoff value of steric and electrostatic descriptors was shortened to its original value (±30 kcal/mol), and the scale was set to the CoMFA standard.
 | Table 1. Structure of 1,6-naphthyridine and pyridopyrimidine analogues and their inhibitory activity against VEGFR-2. [Click here to view] |
In CoMSIA, the binding capacities of the molecule were related to the alterations in the molecular feature exposed by the field, which is predominantly an extension of CoMFA. At all grid points, the distance of the probe atom and molecular atom was calculated using the Gaussian function. The calculation for CoMSIA is as follow:
Where A represents the similarity index at grid point q, summed over all atoms i of the molecule j under investigation. Wprobe,k is the probe atom with radius 1 Å, charge +1, hydrophobicity +1, hydrogen bond donating +1, hydrogen bond accepting +1. wik is the actual value of the physicochemical property k of atom i. riq is the mutual distance between the probe atom at grid point q and atom i of the test molecule. α is the attenuation factor, with a default value of 0.3, and an optimal value normally between 0.2 and 0.4 (Bhansali and Kulkarni, 2014).
 | Figure 3. Common fragment for the alignment of the data set. [Click here to view] |
 | Figure 4. The aligned data set for CoMFA and CoMSIA) model generation. [Click here to view] |
Partial least square analysis and validation of the model
Partial least square (PLS; an extension of multiple linear regression analysis) was used to generate 3D-QSAR models. If the quantitative structure–activity relationships (QSAR) models are generated with the optimum number of components (ONC) having a higher value of leave-one-out (LOO) cross-validation coefficient (q2) and lower standard error of estimate (SEE), then the probabilities of overfitted models are negligible. Initially, LOO and cross-validation correlation methods (leave half out) were used to determine the predictability of the developed models (q2 and r2cv). The developed models are accepted if the value of q2 > 0.5 and r2 > 0.616 (Golbraikh and Tropsha, 2002). The following equation was used for the calculation of q2:
Conventional correlation coefficient (r2), SEE, and Fisher’s value (F value) were calculated using a non-cross validation method. Then, a bootstrap analysis was carried out (10 cycles; 100 times; r2bs) to check the robustness of the generated models (Raichurkar and Kulkarni, 2003). Lastly, the predictability of generated models was assessed by a test set analogue using the equation:
Where SD is the sum of squared deviations between the inhibitory activity of the test set and the mean activity of the training set analogues, and PRESS is the sum of squared deviations between actual and predicted activity values for each analogue in the test set (Caballero et al., 2010; Kharkar et al., 2002).
Pharmacokinetic (ADME) properties prediction of designed analogues
Pharmacokinetic (ADME) parameters are one of the primary reasons behind the withdrawal of anticancer agents from the market. Furthermore, the adverse drug reactions are dependent directly or indirectly on the pharmacokinetic profile of the drug. In-silico ADME prediction supports the lead optimization process to prevent the withdrawal of drug candidates from the clinical trials. The prediction of pharmacokinetic parameters of designed analogues was carried out using QikProp (Schrödinger, New York, NY). QikProp predicts both physicochemical and pharmaceutical properties and provides ranges by comparing the property of molecules with 95% of known drugs (Dagan-Wiener et al., 2017).
RESULTS AND DISCUSSION
CoMFA and CoMSIA model generation
Electrostatic and steric descriptors were used for the generation of the CoMFA model. Initially, the PLS analysis was performed by “LOO” cross-validation correlation (q2 = 0.659; ONC = 6). Non-cross-validation correlation was done by column filtering 2.0 and the same ONC (r2ncv = 0.987; F value = 439.868; SEE = 0.123). The steric and electrostatic field contributions obtained 5.921 and 0.000, respectively (Table 2). The bootstrap analysis r2bs (0.994) and cross-validation coefficient r2cv (0.621) supported the consistency of the developed QSAR model. The pIC50 values (experimental and predicted) of the data set obtained from the CoMFA model are given in Table 3. and 4. The correlation between actual and predicted activities of training and test set analogues based on the CoMFA model is shown in Figure 5.
 | Table 2. Summary of CoMFA and CoMSIA models. [Click here to view] |
Based on the CoMFA results, CoMSIA was carried out using a similar data set. The cross-validation correlation coefficient (q2) was found to be 0.689, and non-cross validation correlation coefficient (r2ncv) was found to be 0.985 with six ONC. The contribution of CoMFA descriptors are as follows: steric (0.759), electrostatic (0.000), hydrophobic (0.349), H-bond donor (0.189), and H-bond acceptor (0.310). The internal reliability of the data set was evaluated using cross-validation method (r2cv = 0.678). The robustness of the model was determined by bootstrap analysis r2bs (0.992) and SEEbs (0.096) (Table 2). The pIC50 values (actual and predicted) of training and test set analogues based on the CoMSIA model are presented in Table 3 and 4. The correlation between actual and predicted activities of the training and the test set analogues based on the CoMSIA model is shown in Figure 6. The residual activities differences of analogues as histogram based on the CoMFA and CoMSIA model are shown in Figure 7.
3D-QSAR visualization
CoMFA contour maps
3D contour maps are the essential features of the CoMFA and CoMSIA. Contour maps were generated as a result of alternations in the molecular fields (standard deviations and the least squares coefficients; StDev*Coeff) allotted to every single grid intersection within the active site. Based on these contour maps, the chemical structures are modified to optimize the inhibitory activity. The most potent analogue 20 (IC50 = 0.003 µM) and least active analogue 45 were chosen for the analysis of contour maps. Here, around the molecule, a transparent style was selected to visualize the generated contour maps. The steric field is characterized by green (favorable) and yellow contour (unfavorable). In green contour, the incorporation of bulky substituents lead to the enhancement of inhibitory activity, whereas in yellow contour, the incorporation of bulky substituent lead to the loss of biological activity. For better understanding, the potent analogue 20 alienated into three regions: “a,” “b,” and “c” (Fig. 8).
The green contour in Figure 9a at the “b” region suggests that the bulky substituent and substituted urea moieties may be favorable for inhibitory activity. This is evident from the experimental IC50 values (µM) of analogues 14 (0.003), 17 (0.005), 19 (0.006), 20 (0.003), 23 (0.006), 26 (0.004), 102 (0.062), 103 (0.021), and 105 (0.071). In these analogues, -NHCONHtBu, the bulky substituent present at the second position of 1,6-naphthyridine ring and pyridopyrimidine ring, leads to potent inhibitory activity. Whereas in analogues 15 (IC50 = 0.18 µM), 21 (IC50 = 0.22 µM), 27 (IC50 = 2.9 µM), 30 (IC50 = 3.6 µM), and 35 (IC50 = 1.2 µM), -NH2, substituent present at the second position of 1,6-naphthyridine ring, leads to poor efficacy against VEGFR-2. Furthermore, as observed in Figure 9a, the yellow contour at “a” region at -NH group suggests that the bulky substituents are unfavorable for inhibitory activity, which is apparent from the actual IC50 values (µM) of analogues 39 (0.67), 40 (0.65), 44 (1.9), 45 (13), and 46 (7.6), where -N(Ac)(CH2)xOBn bulky substituent present at the seventh position of 1,6-naphthyridine ring leads to a remarkable loss of biological activity. Furthermore, the green contours were observed at the phenyl ring (third position of 1,6-naphthyridine and pyridopyrimidine ring), which indicates that phenyl ring substituted with bulky groups is favorable for inhibitory activity. Here, phenyl ring substituted with 3,5-diOMe and 2,6-diCl substituents exhibits potent activity in comparison to the unsubstituted phenyl ring. This is apparent from the experimental IC50 values (µM) of analogues 27 (2.9), 29 (8.7), 30 (3.6), and 31 (0.18).
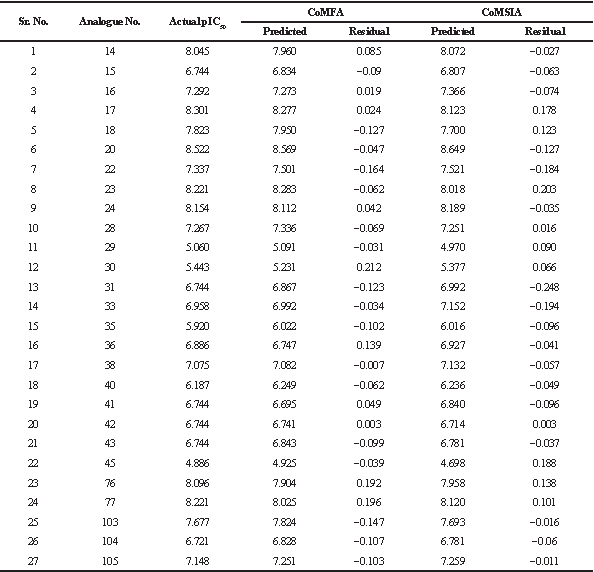 | Table 3. Actual and predicted pIC50 values and residual values of training set analogues by CoMFA and CoMSIA. [Click here to view] |
 | Table 4. Actual and predicted pIC50 values and residual values of test set analogues by CoMFA and CoMSIA. [Click here to view] |
 | Figure 5. The plot of actual and predicted activities of the training and the test set analogues based on the CoMFA model. [Click here to view] |
 | Figure 6. The plot of actual and predicted activities of the training and the test set analogues based on the CoMSIA model. [Click here to view] |
Comparative Molecular Similarity Indices Analysis contour maps
CoMSIA steric field contour maps are almost homogenous to CoMFA. Additionally, in CoMSIA steric map, the green contour at the “c” region in Figure 10a indicates that the bulky substituents favored the potent activity. In most of the analogues, the 1,3-dimethoxyphenyl ring present at the third position of 1,6-naphthyridines and pyridopyrimidines suggests that the replacement with more bulky groups may enhance the VEGFR-2 inhibitory activity. Furthermore, similar to CoMFA steric maps, the yellow contour was observed at the “a” region specifically at -NH group, which suggests that the tertiary amine group is unfavorable for the activity. This is apparent from the IC50 values of analogues 39, 40, 44, 45, and 46, where -N(Ac)(CH2)xOBn substituent present which results in a remarkable loss of activity. Additionally, at “a” region green contour near aliphatic linker indicates that bulky substituents are favored in this region. The generated contour maps from CoMSIA for the most active analogue 20 and the least active analogue 45 represented in Figures 10 and 11.
The hydrophobic field are represented by yellow and white contour maps (Fig. 10b). To enhance the inhibitory activity, hydrophobic groups are favorable in the yellow contour, while hydrophilic groups are favorable in white contour. In Figure 10b at “c’ region near phenyl ring, a large yellow contour suggests that hydrophobic substituent can be favorable for inhibitory activity. While in “a” and “b” region, white contour indicates that the hydrophilic group can enhance the activity.
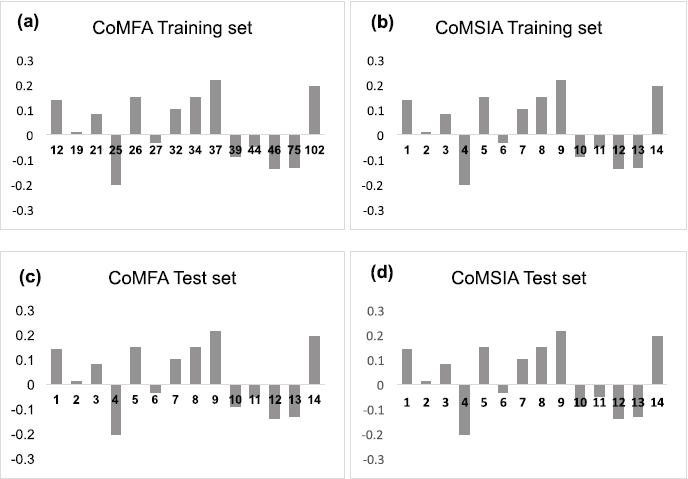 | Figure 7. (a, b) Histogram of CoMFA and CoMSIA residual values for the training set analogues; (c, d) Histogram of CoMFA and CoMSIA residual values for the test set analogues. [Click here to view] |
 | Figure 8. The most potent analogue, 20 alienated into (a), (b), and (c) regions. [Click here to view] |
The CoMSIA donor field is indicated by cyan and purple contour maps. H-bond donor groups are favorable in cyan contour maps. In contrast, purple contour indicates that the H-bond donor groups are unfavorable. Here, in Figure 10c at the “b” region, H-bond donor groups are unfavorable for the biological activity. The acceptor field is characterized by magenta and red contour maps. Magenta contour represents a H-bond acceptor substituent that is favorable, while red contour represents a H-bond acceptor substituent that is not favorable. In Figure 10d, at “b” region magenta contour present at -C=O functional group, which suggests that urea or amide moiety is necessary for the significant biological activity. Replacement of urea functional group with amine leads to loss of activity i.e., analogues 15 (IC50 = 0.18 µM), 21 (IC50 = 0.22 µM), 27 (IC50 = 2.9 µM), and 35 (IC50 = 1.2 µM). While red contour observed at the “b” region indicates that acceptor substituents are unfavoured for inhibitory activity. This is evident from the experimental IC50 values of analogues 39, 40, 44, 45, and 46.
Designing of novel analogues based on the generated 3D-QSAR model
The SAR and structural requirements for inhibition of VEGFR-2 were identified using the analysis of contour maps (Fig. 12). After studying SAR, a novel molecules were designed and aligned to the previously generated data set and the activity was predicted based on the CoMSIA model. Structures and predicted pIC50 values of designed analogues are reported in Table 5. The majority of compounds exhibit pIC50 values are equivalent to analogue 20, which strongly suggests that the generated 3D-QSAR model is valid for designing novel ligands against VEGFR-2.
ADME Studies
The in silico prediction of ADME properties identifies whether the new molecules could be toxic or nontoxic, able or unable to cross membranes, and can be metabolized by the body into an active or inactive form. ADME predictions using QikProp involves the use of molecular descriptors. The pharmaceutically relevant descriptors are shown in Table 6. Log P represents lipophilicity, which is a crucial factor governing passive membrane partitioning. Increased log P value increases permeability while reducing solubility. The octanol–water partition coefficient (QPlogPo/w) and aqueous solubility (QPlogS) were found in the range of 3.140–5.056 and –6.227 to –3.923, respectively. The blood-brain barrier (BBB) partition coefficient (QPlogBB) was found ranging from –0.019 to 0.269. QPlogHERG (predicted IC50 value for the blockage of human Ether-à-go-go-Related Gene (HERG) K+ channels) ranged from –8.144 to –6.479. QPPCaco (Caco-2 cell permeability; mm/second) and QPlogKhsa (Prediction of binding to human serum albumin) varied from 12.733 to 170.97 and 1.020 to 0.328, respectively. Moreover, the percentage of human oral absorption was found in the range of 39.190%–88.895% for all the analogues. The pharmacokinetic parameters for the 16 designed analogues fell under the acceptable range that can be suitable for human use, which also reveals the potential of designed analogues as possible drug-like candidates.
 | Figure 9. Contour maps of CoMFA (StDev*Coeff) (a, b) steric field for analogue 20 and analogue 45. StDev*Coeff, standard deviations, and the least-squares coefficients allotted to each grid intersection were contoured within the binding pocket [Click here to view] |
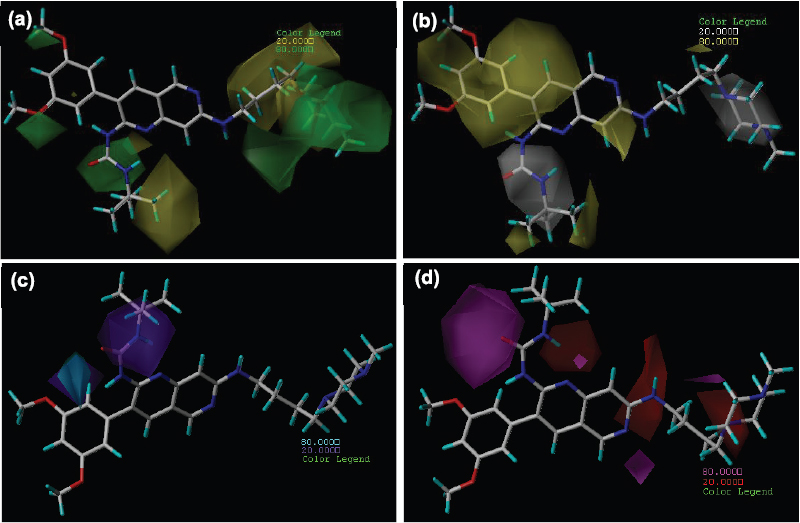 | Figure 10. Contour maps of CoMSIA (stDev*Coeff) (a) steric (b) hydrophobic (c) H-bond donor and (d) H-bond acceptor fields for analogue 20. StDev*Coeff, standard deviations, and the least-squares coefficients assigned to each grid intersection were contoured within the binding pocket. [Click here to view] |
 | Figure 11. Contour maps CoMSIA (stDev*Coeff) (a) steric (b) hydrophobic (c) H-bond donor and (d) H-bond acceptor fields for analogue 45. StDev*Coeff, standard deviations, and the least-squares coefficients assigned to each grid intersection were contoured within the binding pocket. [Click here to view] |
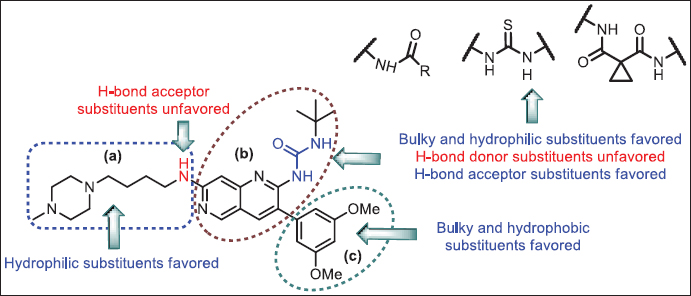 | Figure 12. SAR based on 3D-QSAR. [Click here to view] |
 | Table 5. Predicted pIC50 values of designed analogues using the developed CoMSIA model. [Click here to view] |
 | Table 6. ADME properties of designed analogues. [Click here to view] |
CONCLUSION
VEGFR-2/KDR is a transmembrane receptor tyrosine kinase expressed on the endothelial cells and is activated by VEGFs, which is a promising target for angiogenesis inhibition and suppresses tumor growth. We performed 3D-QSAR using CoMFA and CoMSIA models to correlate the structural parameters of 1,6-naphthyridine and pyridopyrimidine analogues with their VEGFR-2 inhibitory activity. PLS analysis was carried out to evaluate the developed 3D-QSAR model, which indicates that all statistical parameters were obtained reasonably. The conventional correlation coefficient (r2ncv) and cross-validation coefficient (q2) were found to be 0.985 and 0.987 and 0.659 and 0.689 for CoMFA and CoMSIA, respectively. The predictability of CoMSIA was found to be better than CoMFA. The results suggest that steric, hydrophobic, donor and acceptor descriptors have a remarkable impact on inhibitory activity; on the contrary, the electrostatic field has no contribution. In 1,6-naphthyridine analogues, the substituents present on the 3-phenyl ring are significant for inhibitory activity. The order of inhibitory activity was found as follows: (3-(3,5-dimethoxyphenyl) > 3-(2,6-dichlorophenyl) > 3-phenyl). The contour maps analysis suggests that bulky, hydrophobic and H-bond acceptor substituents are favored at the second position of 1,6-naphthyridine ring near the “b” region. The replacement of urea moieties in analogues with thiourea or amide may enhance the biological activity. Furthermore, at the third position of 1,6-naphthyridine ring at the “c” region, the bulky and hydrophobic substituent could enhance the biological activity. Moreover, majority of FDA approved VEGFR-2 inhibitors consist of (I) aromatic ring or nitrogen-containing heteroaromatic ring, (II) core aromatic or heteroaromatic ring either monocyclic or bicyclic, (III) amide/urea/thiourea functional group (H-bond acceptor and H-bond donor group), (IV) an aromatic ring substituted with halogens. On the same line, the reported 1,6-naphthyridine and pyridopyrimidine analogues also consist of an aromatic ring attached via an aliphatic linker, core nitrogen-containing aromatic bicyclic ring, urea functional group, and substituted aromatic ring. Based on the developed 3D-QSAR model, contour map analysis, and structure finding, novel inhibitors were designed with little modification on the most potent analogue 20, and their activities were predicted using the CoMSIA model. The ADME properties of designed analogues were also found to be satisfactory in order to become drug-like candidates. The synthesis and biological activity screening of designed compounds were undertaken. The 3D-QSAR analysis summarised in this research work can be useful for the designing of novel 1,6-naphthyridine and pyridopyrimidine analogues as VEGFR-2 inhibitors.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Shivajirao Kadam, Chancellor, Bharati Vidyapeeth (Deemed to be University), Pune, and Dr. Kakasaheb R. Mahadik, Principal, Poona College of Pharmacy, Pune, Maharashtra, India, for their encouragement.
AUTHOR CONTRIBUTIONS
The authors have approved the final version of the manuscript and contributed equally.
CONFLICT OF INTEREST
The authors declare no conflict of interest pertaining to this manuscript.
FUNDING
There was no funding source pertaining to this research work.
REFERENCES
Abdullah SE, Perez-Soler R. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer, 2012; 118:3455–67. CrossRef
Allergan, Inc. US20160096832. Preparation of 7,8-dihydropyrido[4,3-d]pyrimidine derivatives as tyrosine kinase inhibitors. 2016.
Allergan, Inc. US20160096837. Preparation of pyrazolo[1,5-a]pyrido[3,4-e]pyrimidine derivatives as tyrosine kinase inhibitors. 2016.
Basu A, Jasu K, Jayaprakash V, Mishra N, Ojha P, Bhattacharya S. Development of CoMFA and CoMSIA models of cytotoxicity data of anti-HIV-1-phenylamino-1H-imidazole derivatives. Eur J Med Chem, 2009; 44:2400–7. CrossRef
Bhansali SG, Kulkarni VM. Combined 2D and 3D-QSAR, molecular modelling and docking studies of pyrazolodiazepinones as novel phosphodiesterase 2 inhibitors. SAR QSAR Environ Res, 2014; 25:905–37. CrossRef
Caballero J, Fernández M, Coll D. Quantitative structure–activity relationship of organosulphur compounds as soybean 15-Lipoxygenase inhibitors using CoMFA and CoMSIA. Chem Biol Drug Des, 2010; 76:511–7. CrossRef
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature, 2011; 473:298–307. CrossRef
Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc, 1988; 110:5959–67. CrossRef
Dagan-Wiener A, Nissim I, Abu N Ben, Borgonovo G, Bassoli A, Niv MY. Bitter or not? BitterPredict, a tool for predicting taste from chemical structure. Sci Rep, 2017; 7:1–13. CrossRef
Ellis LM, Hicklin DJ. Pathways mediating resistance to vascular endothelial growth factor–targeted therapy. Clin Cancer Res, 2008; 14:6371–5. CrossRef
Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res, 2017; 7:1016.
Frampton JE. Vandetanib. Drugs, 2012; 72:1423–36. CrossRef
Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Model, 2002; 20:269–76. CrossRef
Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, Epperly AH, Hinkle KW, Hunter RN, Johnson JH, Knick VB, Laudeman CP, Luttrell DK, Mook RA, Nolte RT, Rudolph SK, Szewczyk JR, Truesdale AT, Veal JM, Wang L, Stafford JA. Discovery of 5-[[4-[(2, 3-dimethyl-2 H-indazol-6-yl) methylamino]-2-pyrimidinyl] amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem, 2008; 51:4632–40. CrossRef
Ho TH, Jonasch E. Axitinib in the treatment of metastatic renal cell carcinoma. Future Oncol, 2011; 7:1247–53. CrossRef
Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet, 2016; 388:518–29. CrossRef
Kharkar PS, Desai B, Gaveria H, Varu B, Loriya R, Naliapara Y, Shah A, Kulkarni VM. Three-dimensional quantitative structure-activity relationship of 1, 4-dihydropyridines as antitubercular agents. J Med Chem, 2002; 45:4858–67. CrossRef
Kiselev E, Dexheimer TS, Pommier Y, Cushman M. Design, synthesis, and evaluation of dibenzo [c, h][1, 6] naphthyridines as topoisomerase I inhibitors and potential anticancer agents. J Med Chem, 2010; 53(24):8716–26. CrossRef
Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem, 1994; 37:4130–46. CrossRef
Kumar R, Crouthamel MC, Rominger DH, Gontarek RR, Tummino PJ, Levin RA, King AG. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer, 2009; 101:1717–23. CrossRef
Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov, 2019; 2:100009; doi:10.1016/j.medidd.2019.100009. CrossRef
Raichurkar AV, Kulkarni VM. Understanding the antitumor activity of novel hydroxysemicarbazide derivatives as ribonucleotide reductase inhibitors using CoMFA and CoMSIA. J Med Chem, 2003; 46:4419–27. CrossRef
Roskoski Jr R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun, 2007; 356:323–8. CrossRef
Salerno S, Barresi E, García-Argáez AN, Taliani S, Simorini F, Amendola G, Tomassi S, Cosconati S, Novellino E, Da Settimo F, Marini AM. Discovery of Pyrido [3′, 2′: 5, 6] thiopyrano [4, 3-d] pyrimidine-based antiproliferative multikinase inhibitors. ACS Med Chem Lett, 2019; 10(4):457–62. CrossRef
Shepard DR, Garcia JA. Toxicity associated with the long-term use of targeted therapies in patients with advanced renal cell carcinoma. Expert Rev Anticancer Ther, 2009; 9:795–805. CrossRef
SYBYL X Molecular Modeling Software. Tripos Associates, V. 2.0. St Louis, USA, 2012. Available via http://www.tripos.com
Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci, 2005; 109:227–41. CrossRef
Thompson AM, Delaney AM, Hamby JM, Schroeder MC, Spoon TA, Crean SM, Hollis Showalter HD, Denny WA. Synthesis and structure– activity relationships of soluble 7-substituted 3-(3, 5-dimethoxyphenyl)-1, 6-naphthyridin-2-amines and related ureas as dual inhibitors of the fibroblast growth factor receptor-1 and vascular endothelial growth factor receptor-2 Tyrosine kinases. J Med Chem, 2005; 48:4628–53. CrossRef
Wang MS, Zhuo LS, Yang FP, Wang WJ, Huang W, Yang GF. Synthesis and biological evaluation of new MET inhibitors with 1, 6-naphthyridinone scaffold. Eur J Med Chem, 2020; 185:111803. CrossRef
Woo HY, Heo J. Sorafenib in liver cancer. Expert Opin Pharmacother, 2012; 13:1059–67. CrossRef
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou Y-C, Joly AH. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther, 2011; 10:2298–308. CrossRef
Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist, 2015; 20:660. CrossRef
Zhu W, Chen G, Hu L, Luo X, Gui C, Luo C, MokPuah C, Chen K, Jiang H. QSAR analyses on ginkgolides and their analogues using CoMFA, CoMSIA, and HQSAR. Bioorg Med Chem, 2005; 13:313–22. CrossRef