
INTRODUCTION
Atherosclerosis is the most important pathophysiological process that leads to cardiovascular diseases (CVD), gangrene, and stroke (Galkina and Ley, 2009). The mortality and morbidity caused by atherosclerotic-led circulatory diseases worldwide are about 31.9% and 54.1% of all deaths in 2010 and 2011, respectively, in the United States alone National Center for Health Statistics (2015), and its cost is about USD315.4 billion in 2010 alone (Go et al., 2014). Atherosclerosis is a complex disease that resulted through various stages and involved many components of the metabolic, immune, and vascular systems (Galkina and Ley, 2009). Atherosclerosis takes place in the subendothelial space of medium and large arteries at regions of disturbed flow of blood. It is prompted by an interaction between the dysfunction of the endothelial lining of blood vessels, which rises its permeability and modification of high levels of cholesterols that are carried in the form of low-density lipoprotein-cholesterols (LDL-C) (Tabas et al., 2015). Later, the LDL-C is oxidized and transported from lumen to subendothelial layer of blood vessels. This will trigger an inflammatory response and formation of macrophages, which then accumulate more and more LDL-C to form lipid-loaded foam cells, the major form of the atherosclerotic lesion (Maiolino et al., 2013). Fibrous caps then form after the stimulation and recruitment of smooth muscle and other inflammatory cells and further deposition of lipid materials (Bentzon et al., 2014). This prolonged process may develop fibrous plaques. At this stage, the occlusive lesions contain a dense cap of fibrous connective tissues, which comprise smooth muscle cells, macrophages, and T-lymphocytes, and are surrounded by the dense layers of connective tissue matrix containing collagen as the major component and less amount of elastic fibers. In an advanced stage, the size of the fibrous plaques will become larger and will intrude into the arterial lumen which then blocks the flow of blood and oxygen to the affected part and causes devastating clinical consequences, including cerebral infarction or stroke in the brain, myocardial infarctions, and loss of function in the peripheral vascular system (Tabas et al., 2015). Therefore, a high level of plasma LDL-C is the most critical influence in the commencement and progression of atherosclerosis (Steinberg and Witztum, 2010). The LDL-receptor (LDL-R), which presents on the cell surface of liver cells, regulates the level of plasma LDL-C. The inverse relationship between LDL-R and LDL-C is well discussed. LDL-R is one of the genes, which is associated with familial hypercholesterolemia (FH) (Goldstein and Brown, 2009). It was discovered that various mutations of the LDL-R occurred in patients with FH. LDL-R functions by the binding of LDL-C to the receptor on the cell membrane of hepatocytes, which triggers the process of internalization of the complex of LDL-R and LDL-C into the cells via endocytosis. In lysosome, due to pH regulation, LDL-R is dissociated from LDL-C and recycled back to the cell surface to continue its function in removing the excess LDL-C from circulation (Tomkin and Owens, 2012).
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a serine protease and synthesized in the liver. It can be either retained in the cytoplasm or secreted into the blood where it plays a key role in regulating the life cycle and the number of LDL-R (Catapano and Papadopoulos, 2013; Seidah et al., 2014). It is the third gene known to be associated with autosomal dominant familial hypercholesterolemia after apoB100 and LDL-R (Cariou et al., 2011). In a normal situation, on the binding of LDL-R to LDL-C, the complex is brought up into the liver cells by endocytosis. After releasing LDL-C, LDL-R then returns to the cell surface. Here again, the receptors bind with another circulating LDL-C particles. However, the secreted PCSK9 can bind to LDL-R and it targets LDL-R for lysosomal degradation, thus preventing the receptor from recycling back to the cell surface. PCSK9 enzyme also enhances the degradation of LDL-R intracellularly through a partly defined intracellular pathway (Poirier et al., 2009). Thus, PCSK9 leads to a reduction in the number of LDL–R existing on the cell surface of hepatocytes. Thus, the LDL-C level will be elevated initiating the development of atherosclerosis. Therefore, the inhibition of PCSK9 activity offers a promising therapeutic target to reduce the plasma level of LDL-C which still stands as the basis in the atherosclerosis treatment (Verbeek et al., 2015).
Currently, statins are widely used to reduce the risk of atherosclerosis. Statins are the inhibitors for 3-hydroxy-3-methylglutaryl-CoA reductase, an enzyme for transforming HMG-CoA into mevalonic acid which is a precursor for cholesterol synthesis in the liver (Rubinstein and Izkhakov, 2002). Despite a significant role of statins to lower the synthesis of cholesterol in the liver which reduces the level of circulating LDL--C, burden of atherosclerosis and CVD diseases is still high (Verbeek et al., 2015). Statins are effective at helping to reduce the cholesterol level and reduce the risk of heart problems, but they can cause side effects for some people. Several common side effects are headache, pins and needles, abdominal pain, bloating, diarrhea, feeling sick, and rash. Some statin drugs may impair memory, and it was demonstrated that two commonly prescribed statins, pravastatin (Pravachol) and atorvastatin (Lipitor), reduced the performance of recognition and working memory in an animal experiment. Taking statins has been linked with a reduction in CoQ10 level in the body, which may protect cells from damage, affect blood sugar levels, and increase the risk of diabetes (Aiman et al., 2014; Richard-Deichmann et al., 2010). Hepatocyte LDL-R number increases on statin treatment, but at the same time, the plasma level of PCSK9 also elevates; thus, the latter decreases the uptake of LDL-C by the liver cells and increases plasma LDL-C, diminishing the efficiency of statins. In addition, several studies showed that despite reducing the levels of cholesterols in the liver cells, statins also activate the transcription factors such as SREBP which binds to the promoter region of PCSK9 and increases the level of the enzyme (Jia et al., 2014). This phenomenon results in the degradation of LDL-R protein and elevates the level of LDL (Li et al., 2009). It was reported that 2- to 3-fold increase in LDL-R was observed and a corresponding 25%–50% reduction in circulating cholesterol in PCSK9 knockout mice (Rashid et al., 2005), indicating that PCSK9 performs an important new target in the discovery of new drugs for therapeutic intervention against atherosclerosis.
Three approaches targeted different phases of PCSK9 synthesis or function have been used and undergone preclinical/clinical trials (Stock, 2015; Verbeek et al., 2015). The first approach involves the usage of antisense oligonucleotides and small interfering ribonucleic acids to decrease the synthesis of PCSK9 protein by the degradation of PCSK9 mRNA (Fitzgerald et al., 2014; Lindholm et al., 2012). The second approach involves the development of agents that interfere with the binding of PCSK9 to LDL–R, which includes adnectins, monoclonal antibodies, and small peptides that can prevent the degradation of LDL-R. Among them, monoclonal antibodies (evolocumab and alirocumab) are now in the late-stage (phase 3 clinical trials) testing. The use of both evolocumab and alirocumab is the most efficient as it reduces the level of circulating LDL by 16%–19% and 47%–57% compared to placebo, respectively (Koren et al., 2014; Roth et al., 2014). The third approach involves the inhibition of PCSK9 production by targeting its intracellular processing which has not reached clinical development (Lambert et al., 2012). However, all of the approaches do not target PCSK9 at the transcriptional level which is reducing the promoter activity of the gene. It was extensively established that the cis-acting elements present on the PCSK9 promoter are the binding sites for many transcription factors such as HNF1a, SREBP, Peroxisome proliferator-activated receptor gamma (PPARγ), and PPARγ (Dong et al., 2010; Jeong et al., 2008; Li et al., 2009). Therefore, targeting PCSK9 at the transcriptional level by manipulating its promoter will form a significant target to identify and validate marine compounds with potent activity in reducing the expression level of PCSK9 and subsequently increasing the level of LDL–R, reducing the level of LDL, and more importantly reducing the progression of atherosclerosis.
Natural products (terrestrial or marine origins) have been continuously used for the prevention and curing of ailments or diseases. Marine natural products have attracted the attention of scientists worldwide for the past five decades. To date, more than 16,000 marine natural products have been characterized from marine organisms. Every year, hundreds of new natural products are discovered from the marine organisms. Finding new and novel bioactive compounds is one of the important drivers for this field of research (Hu et al., 2015). At present, although the technology is developed, with realization of their chemical diversity, natural products are a better candidate for successful drugs rather than the vast diversity of synthetic compound libraries (Feher and Schmidt, 2003; Grabowski and Schneider, 2007; Luo et al., 2014), and the former remains as the starting materials in drug discovery (Galm and Shen, 2007). Metabolites from the marine organisms comprise a unique skeleton that their terrestrial counterparts are incapable to produce them. Since they have a great diversity of chemical structures, they have various properties for various diseases such as anticancer, anti-inflammatory, antidiabetic, antihypercholesterolemia, and antiatherosclerosis (Blunt et al., 2015; Mayer and Lehmann, 2000; Newman and Cragg, 2007; Proksch et al., 2002). Some studies reported the roles of marine-based bioactive compounds correlated to CVD, especially to atherosclerosis and hypercholesterolemia or hyperlipidemia. Manzamine A, a marine-derived alkaloid which is isolated from marine sponge Acanthostrongylophora ingens, inhibits the accumulation of cholesterol ester in macrophages by inhibiting acyl-cholesterol acyltransferase (ACAT) activity and suppresses hyperlipidemia and atherosclerosis in vivo (Eguchi et al., 2013). Orally administered manzamine A to apolipoprotein E (apoE)-deficient mice for 80 days significantly reduced total cholesterol, free cholesterol, LDL-C, and triglyceride levels in serum, and the area of atherosclerosis lesions in the aortic sinus was also substantially diminished. Moreover, mycoepoxydiene, a novel polyketide, isolated from marine fungus Diaporhte sp. (Xia et al., 2015) reduces the atherosclerotic lesions in ApoE-deficient mice through the suppression of macrophage foam cell formation and expression of lactin-like oxidized low-density lipoprotein. Furthermore, various compounds from Xestospongia may also play an important role in the treatment of CVD through different mechanisms of action; for example, strongylin A, wiedendiol A, and wiendendiol B show the inhibitory activities of the cholesteryl ester transfer protein (Zhou et al., 2010). In addition, lipid-lowering C12 polyketides, cladospolide E, secopatulolides A and C, 11-hydroxy-c-dodecalactone, and iso-cladospolide B were isolated from a soft coral-derived fungus Cladosporium sp. TZP-29 (Zhu et al., 2015).
The preliminary study on aaptaminoids from a marine sponge Aaptos aaptos and methyl benzoate from a starfish Acanthaster planci showed that these compounds increased the expression of SRB1 and PPARγ (Habsah et al., 2017; Mat Lazim et al., 2016). PPARγ is a transcription factor for PCSK9. Some PPAR activators can reduce the synthesis of PCSK9 gene. Thus, in this study, we modified the structure of these two compounds to study its effect on the inhibition of the PCSK9 gene expression, thus reducing the degradation of the LDR receptor that leads to decrease LDL-C in the body system.
MATERIALS AND METHODS
All reactions were carried out under an ambient atmosphere without taking any special precautions to exclude air and moisture during synthetic workup. All chemicals and solvents which were commercially available were used without any further purification. Infrared spectra were recorded from KBr pellets using PerkinElmer 100 spectrometer in the region of 400–4,000 cm-1. 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra using CDCl3 as solvent were generated via Bruker AVANCE III 400 MHz spectrometer at room temperature electron impact mass spectral (EIMS) data for aaptaminoids were determined using JEOL JMS-T100LP spectrometer, whereas for benzamide–indane using Agilent, 7890A GC spectrometer. High Resolution Electrospray Ionization-Mass Spectrometry (HRESI-MS) was determined using a Waters, VION Ion Mobility QTOF mass spectrometer.
Synthesis and characterization of aaptaminoid derivatives
The synthesis of N1,N4-bisbenzylaaptamine (A1) was prepared as reported by Abdul Razak et al. (2015) (Fig. 1). Aaptamine (0.50 g, 2.19 mmol) was dissolved in anhydrous dimethylformamide (50 ml) with potassium carbonate (1.51 g, 10.95 mmol) and alkyl halide (10.95 mmol) added at room temperature. After stirring for 24 hours at the same temperature, the solution was filtered, and the solvent was removed in vacuo. The residue was purified using a column chromatography (DCM:MeOH of 6:1) (Abdul Razak et al., 2015; Pettit et al., 2004).
 | Figure 1: Synthesis of N1, N4-bisbenzylaaptamine (73% yield) [A1]. [Click here to view] |
N1,N4-Bisbenzylaaptamine (A1, Fig. 1) (0.29 g, 73% yield) was obtained by recrystallization from ethyl acetate–methanol mixture via slow evaporation of the solvent. IR (KBr) vmax: 2,980, 2,940, 1,648, 1,569, 1,304, 1,207, 1,060 cm-1; UV (MeOH) λmax (log Æ) 204 (4.67), 244 (4.39), 263 (4.39), 273 (4.33), 307 (3.62), 415 (3.92) nm ;1H NMR (400 MHz) δ 3.60 (s, 3H, OCH3), 4.05 (s, 3H, OCH3), 5.40 (s, 2H, NCH2), 5.74 (s, 2H, NCH2), 6.53 (d, J = 7.6 Hz, 1H, H-3), 7.13 (d, J = 7.6 Hz, 1H, H-6), 7.20 (d, J = 7.2Hz, 2H, CHar), 7.33–7.46 (m, 8H, CHar), 7.30 (s, 1H, H-7), 7.62 (d, J = 8.8 Hz, 1H, H-5), 7.97 (d, J = 7.6 Hz, 1H, H-2); 13C NMR (100 MHz, CD3OD) δ 55.8, 56.4, 60.1, 60.9, 97.7, 102.9, 114.6, 119.1, 125.8, 126.6, 127.5, 128.2, 128.6, 129.0, 132.9, 133.5, 133.8, 134.3, 135.1, 136.5, 149.1, 149.6, 159.0; EIMS m/z [M]+ found 409.25 (calcd C27H25N2O2, 409.50).
The synthesis of N4-[(3,4,5-trimethoxy)benzyl]aaptamine (A2) and N1-[(3,4,5-trimethoxy)benzyl]aaptamine (A3) was prepared by dissolving 200 mg (0.88 mmol) of aaptamine in DMF (20 ml) at 0°C (Fig. 2). Then, approximately three equivalents of KH were added, and the mixture was stirred for 20 minutes, after which 1.2 equivalents of the appropriate alkyl halide were added dropwise. The reaction was stirred for an additional hour and then allowed to warm to room temperature. The reaction was monitored using thin-layer chromatography at room temperature to ensure the complete conversion of the starting material, which occurred usually after 18–24 hours. The workup consisted of aqueous extraction with CHCl3, a brine wash, and dried over Na2SO4 before evaporated under reduced pressure. Final purification was completed on a silica flash column using 5% MeOH in ammonia-saturated dichloromethane (DCM), which in most cases provided two regioisomers that required multiple repetitions of the same chromatography step to purify (Abdul Razak et al., 2015; Bowling et al., 2008).
N4-[(3,4,5-Trimethoxy)benzyl]aaptamine (A2, Fig. 2) (0.156 g, 31% yield) was obtained by N4-alkylation. IR (KBr) vmax: 2,943, 1,651, 1,598, 1,322, 1,247, 1,126 cm-1; UV (MeOH) λmax (log Æ) 205 (4.72), 239 (4.42), 259 (4.35), 397 (4.62) nm; 1H NMR (400MHz) δ 3.63 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.76 (s, 6H, OCH3), 3.97 (s, 3H, OCH3), 5.43 (s, 2H, NCH2), 6.21 (d, J = 7.6 Hz, 1H, H-3), 6.58 (s, 2H, CHar), 6.87 (s, 1H, H-7), 6.90 (d, J = 7.6 Hz, 1H, H-6), 7.39 (d, J = 7.6 Hz, 1H, H-5), 7.55 (d, J = 7.2 Hz, 1H, H-2); 13C NMR (100 MHz, CD3OD) δ 56.6, 56.7, 59.5, 61.1, 61.9, 70.0, 100.6,110.6, 120.1, 134.9, 135.1, 135.6, 137.0, 146.0, 154.9, 159.4; EIMS m/z [M-Cl]+ found 408.30 (cacld C23H24N2O5, 408.45). N1-[(3,4,5-Trimethoxy)benzyl]aaptamine (A3, Fig. 2) (0.12 g, 25% yield) was obtained via N1 alkylation of aaptamine. IR (KBr) vmax: 2,940, 2,840, 1,646, 1,600, 1,335, 1,239, 1,124 cm-1; UV (MeOH) λmax (log Æ) 204 (4.80), 259 (4.34), 277 (4.21), 314 (3.59), 405 (3.83) nm; 1H NMR (400 MHz) δ 3.64 (s, 3H, OCH3), 3.69 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 5.13 (s, 2H, NCH2), 6.39 (d, J = 7.6 Hz, 1H, H-3), 6.58 (s, 2H, CHar), 6.51 (s, 2H, CHar), 6.93 (d, J = 7.6 Hz, 1H, H-6), 7.09 (s, 1H, H-7), 7.39 (d, J = 7.6 Hz, 1H, H-5), 7.78 (d, J = 7.2 Hz, 1H, H-2); 13C NMR (100 MHz, CD3OD) δ 56.6, 56.7, 59.4, 61.1, 61.9, 100.6, 105.0, 115.1, 134.9, 135.0, 146.0, 154.8, 159.3; EIMS m/z [M]+ found 408.25 (cacld C23H24N2O5, 408.45).
 | Figure 2: Synthesis of N4-[(3,4,5-trimethoxyl)benzyl]aaptamine (31% yield) [A2] and N1-[(3,4,5-trimethoxyl)benzyl] aaptamine (25% yield) [A3]. [Click here to view] |
Synthesis and characterization of benzamide–aminoindane derivatives
Benzamides–aminoindane derivatives were synthesized according to the previous method by Wohlfart et al. (2004) with modification. Figure 3 shows the reaction scheme of the procedure.
The synthesis of N-(2,3-dihydro-1H-inden-2-yl)methoxybenzamide (B1) was prepared by mixing 2-aminoindane (0.50 g, 3.75 mmol) and 0.78 g triethylamine (7.70 mmol) with 4 ml of tetrahydrofuran. Then, 3.94 mmol benzoyl chloride was added dropwise, and the mixture was stirred within a period of 2 hours at room temperature. The resulting mixture was subsequently poured onto ice/HCl mixture. The obtained precipitate was filtered, washed, and later dried in vacuo. Methanol and chloroform were used during the recrystallization of the crude product. The percentage yield was 56.77%.
The same procedure was repeated for synthesizing N-(2,3-dihydro-1H-inden-2-yl)-2-methoxybenzamide (B2), N-(2,3-dihydro-1H-inden-2-yl)-3-methoxybenzamide (B3), N-(2,3-dihydro-1H-inden-2-yl)-4-methoxybenzamide (B4), N-(2,3-dihydro-1H-inden-2-yl)-4-fluorobenzamide (B5), N-(2,3-dihydro-1H-inden-2-yl)-4-chlorobenzamide (B6), N-(2,3-dihydro-1H-inden-2-yl)-4-bromobenzamide (B7), N-(2,3-dihydro-1H-inden-2-yl)-4-iodobenzamide (B8), N-(2,3-dihydro-1H-inden-2-yl)-4-cyanobenzamide (B9), N-(2,3-dihydro-1H-inden-2-yl)-4-cinnamamide (B10), and N-(2,3-dihydro-1H-inden-2-yl)-3,4-dimethoxybenzamide (B11) , but here B2 [3- methoxybenzoyl chloride (0.30 ml, 2.37 mmol), B3 [3-methoxybenzoyl chloride (0.30 ml, 2.37 mmol)], B4 [4-methoxybenzoyl chloride (0.32 ml, 2.37 mmol)], B5 [4-fluorobenzoyl chloride (0.28 ml, 2.37 mmol)], B6 [4-chlorobenzoyl chloride (0.57 ml, 2.37 mmol)], B7 [4-bromobenzoyl chloride (0.52 g, 2.37 mmol)], B8 [4-iodobenzoyl chloride (0.63 g, 2.37 mmol)], B9 [2-aminondane hydrochloride (0.30 g, 1.77 mmol) and 4-cyanobenzoyl chloride (0.37 g, 1.86 mmol)], B10 [cinnamoyl chloride (0.21 g, 1.24 mmol)], and B11 [2-aminondane hydrochloride (0.30 g, 1.77 mmol) and 3,4-dimethoxybenzoyl chloride (0.37 g, 1.86 mmol)] were used. The percentage yields are B2 (34.38%), B3 (86.25%), B4 (85.10%), B5 (99.12%), B6 (54.76%), B7 (53.76%), B8 (49.68%), B9 (79.02%.), B10 (93.75%), and B11 (70.83%),
For compounds: N-(2,3-dihydro-1H-inden-2-yl)benzamide (B1) (0.5052 g, 56.77% yield, dark brown solid) (Fig. 3), IR (KBr) vmax: 3,335, 1,633, 1,531, 1,311, 1,228, 1,080 cm-1; 1H NMR (400 MHz) δ 2.93 (dd, J = 16, 4.4 Hz, 2H, H-2), 3.43 (dd, J = 16.2, 7.2 Hz, 2H, H-3), 4.96 (m, 1H, H-1), 7.2 (dd, J = 5.6 ,3.2 Hz, 2H, H-3), 7.25 (t, J = 8.4 Hz, 2H, H-4), 7.40 (t, J = 7.4 Hz, 2H, H-6), 7.48 (t, J = 7.2 Hz, 1H, H-7), 7.72 (d, J = 7.2 Hz, 2H, H-5); 13C NMR (100 MHz, CD3OD) δ 40.3, 51.1, 124.9, 126.9, 126.9, 128.5, 131.5, 134.6, 140.9, 167.3. HRESIMS (positive mode) m/z 238.1231 [M + H]+ (calcd for C16H15NO, 238.2925).
N-(2,3-dihydro-1H-inden-2-yl)-2-methoxybenzamide (B2) (0.3446 g, 34.38% yield, dark brown solid) (Fig. 3), IR (KBr) vmax: 3,394, 1,622, 1,521, 1,293, 1,239, 1,012 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 15.8, 5.2 Hz, 2H, H-2), 3.43 (dd, J = 16, 7.2 Hz, 2H, H-3), 3.82 (s, 3H, OCH3), 4.95 (m, 1H, H-1), 6.92 (d, J = 8.4 Hz, 1H, H-6′), 7.07 (td, J = 7.6, 1.2 Hz, 1H, H-5′), 7.19 (dd, J = 5.6, 3.2 Hz, 2H, H-6, H-9), 7.25 (dd, J = 5.8, 2.4 Hz, 2H, H-7, H-8), 7.42 (td, J = 1.6, 0.8 Hz, 1H, H-4′, 8.2 (dd, J = 7.9, 1.6 Hz, 1H, H-3′); 13C NMR (100 MHz, CD3OD) δ 40.3, 50.9, 55.9, 111.4, 121.4, 121.7, 124.8, 126.7, 132.2, 132.7, 141.2, 157.4, 165.0. HRESIMS (positive mode) m/z 269.1369 [M + 2H]2+ (calcd for C17H17NO2, 269.3159),
 | Figure 3: Synthesis of benzamide derivatives. N-(2,3-Dihydro-lH-inden-2-yl)benzamide (B1) (56.77% yield), N-(2,3-dihydro-lH-inden- 2-yl)-2-methoxybenzamide (B2) (34.38%), N-(2,3-dihydro-lH-inden-2-yl)-3-methoxybenzamide (B3) (86.25%), N-(2,3- dihydro-lH-inden-2-yl)-4-methoxybenzamide (B4) (85.10%), N-(2,3-dihydro-lH-inden-2-yl)-4-fluorobenzamide (B5) (99.12%), N-(2,3-dihydro-lH-inden-2-yl)-4-chlorobenzamide (B6) (54.76%), N-(2,3-dihydro-lH-inden-2-yl)-4-bromobenzamide (B7) (53.76%), N--(2,3-dihydro-lH-inden-2-yl)-4-iodobenzamide (B8) (49.68%), N-(2,3-dihydro-lH-inden-2-yl)-4-cyanobenzamide (B9) (79.02%), N-(2,3-dihydro-lH-inden-2-yl)-4-cinnamamide (B10) (93.75%), N-(2,3-dihydro-lH-inden-2-yl)-3,4- dimethoxybenzamide (B11) (70.83%). [Click here to view] |
N-(2,3-dihydro-1H-inden-2-yl)-3-methoxybenzamide (B3) (0.5188 g, 86.25% yield, white solid) (Fig. 3), IR (KBr) vmax: 3,239, 1,634, 1,547, 1,323, 1,247, 1,034 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16, 4.4 Hz, 2H, H-2), 3.42 (dd, J = 16, 7.2 Hz, 2H, H-3), 3.83 (s, 3H, OCH3), 4.94 (m, 1H, H-1), 6.99 (dd, J = 7.8, 1.2 Hz, 1H, H-6), 7.02 (dd, J = 7.8, 0.8 Hz, 1H, H-9), 7.2 (dd, J = 5.6, 3.3 Hz, 2H, H-7, H-8), 7.25 (dd, J = 7.8, 4.4 Hz, 2H, H-6′), 7.29 (dd, J = 7.8,1.2 Hz, 1H, H-4′), 7.31 (s, 1H, H-2′),7.34 (t, J = 2.0, 2.0 Hz, 1H, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.2, 51.1, 55.5, 112.4, 117.6, 118.6, 124.9, 126.9, 129.5, 136.1, 140.8, 159.8, 167.2. HRESIMS (positive mode) m/z 269.1364 [M + 2H]2+ (calcd for C17H17NO2, 269.3159),
N-(2,3-dihydro-1H-inden-2-yl)-4-methoxybenzamide (B4) (0.5119 g, 85.10% yield, light yellow solid) (Fig. 3), IR (KBr) vmax: 3,329, 1,632, 1,545, 1,294, 1,252, 1,026 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16.4, 4.4 Hz, 2H, H-2), 3.41 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 3.83 (s, 3H, OCH3), 4.94 (m, 1H, H-1), 6.89 (dd, J = 6.8, 2.0 Hz, 2H, H-6, H-9), 7.19 (dd, J = 8.8, 5.2 Hz, 2H, H-7, H-8), 7.25 (d, J = 7.8 Hz, 2H, H-2′, H-6′), 7.7 (d, J = 9.2 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.3, 51.0, 55.4, 113.7, 124.9, 124.9, 126.8, 128.7, 140.9 , 162.1, 166.8. HRESIMS (positive mode) m/z 269.1368 [M + 2H]2+ (calcd for C17H17NO2, 269.3159),
N-(2,3-dihydro-1H-inden-2-yl)-4-fluorobenzamide (B5) (0.4519 g, 94.93% yield, dark brown solid) (Fig. 3), IR (KBr) vmax: 3,288, 1,633, 1,505, 1,360, 1,289, 1,227 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16, 4.4 Hz, 2H, H-2), 3.41 (dd, J = 16, 6.8 Hz, 2H, H-3), 4.93 (m, 1H, H-1), 7.07 (t, J = 8.4 Hz, 2H, H-7, H-8), 7.2 (d, J = 8.4 Hz, 2H, H-6, H-9), 7.25 (d, J = 8.4 Hz, 2H, H-2;, H-6′), 7.74 (dd, J = 8.4, 5.2 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.3 , 51.2, 115.4, 124.9, 126.9, 129.2, 130.7, 140.8, 163.5, 166.3. HRESIMS (positive mode) m/z 256.1181 [M + H]+ (calcd for C16H14NOF, 256.2831),
N-(2,3-dihydro-1H-inden-2-yl)-4-chlorobenzamide (B6) (0.3348 g, 54.76% yield, dark brown solid) (Fig. 3), IR (KBr) vmax: 3,284, 1,632, 1,543, 1,358, 1,203, 1,068 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16, 4.4 Hz, 2H, H-2), 3.4 (dd, J = 16, 6.8 Hz, 2H, H-3), 4.92 (m, 1H, H-1), 7.2 (dd, J = 5.2, 2.8 Hz, 2H, H-7, H-8), 7.25 (d, J = 6.4 Hz, 2H, H-6, H-9), 7.36 (d, J = 8.4 Hz, 2H, H-2′, H-6′), 7.66 (d, J = 8.8 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.2, 51.2, 124.9, 126.9, 128.4, 128.8, 132.9, 137.7, 140.7, 166.2. HRESIMS (positive mode) m/z 272.0837 [M + H]+ (calcd for C16H14NOCl, 272.7377),
N-(2,3-dihydro-1H-inden-2-yl)-4-bromobenzamide (B7) (0.3825 g, 53.76% yield, yellow solid) (Fig. 3), IR (KBr) vmax: 3,280, 1,632, 1,542, 1,424, 1,180, 1,066 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16.4, 4.0 Hz, 2H, H-2), 3.42 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 4.94 (m, 1H, H-1), 7.2 (dd, J = 5.6, 3.2 Hz, 2H, H-7, H-8), 7.25 (d, J = 3.6 Hz, 2H, H-6, H-9), 7.53 (d, J = 8.8 Hz, 2H, H-2′, H-6′), 7.59 (d, J = 8.4 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.3, 51.2, 124.9, 126.1, 126.9, 128.5, 131.8, 133.4, 140.7, 166.2. EIMS m/z 316.2 C16H14NOBr, 316.1887),
N-(2,3-dihydro-1H-inden-2-yl)-4-iodobenzamide (B8) (0.4060 g, 49.68% yield, black solid) (Fig. 3), IR (KBr) vmax: 3,280, 1,631, 1,537, 1,359, 1,302, 1,148 cm-1; 1H NMR (400 MHz) δ 2.92 (dd, J = 16, 4.0 Hz, 2H, H-2), 3.42 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 4.93 (m, 1H, H-1), 7.2 (dd, J = 5.6, 2.8 Hz, 2H, H-7, H-8), 7.26 (d, J = 4.4 Hz, 2H, H-6, H-9), 7.45 (d, J = 8.4 Hz, 2H, H-2′, H-6′), 7.75 (d, J = 8.8 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.3, 51.1, 89.3, 124.9, 126.9, 128.5, 133.9, 137.8, 140.7, 166.5. HRESIMS (positive mode) m/z 366.1399 [M + 3H]3+ (calcd for C16H14NOI, 366.1892),
N-(2,3-dihydro-1H-inden-2-yl)-4-cyanobenzamide (B9) (0.3669 g, 79.02% yield, white solid) (Fig. 3), IR (KBr) vmax: 3,265, 1,621, 1,547, 1,315, 1,287, 864 cm-1; 1H NMR (400 MHz) δ 2.93 (dd, J = 16, 4.0 Hz, 2H, H-2), 3.42 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 4.93 (m, 1H, H-1), 7.2 (dd, J = 7.6, 2.8 Hz, 2H, H-7, H-8), 7.26 (dd, J = 7.6, 2.4 Hz, 2H, H-6, H-9), 7.68 (d, J = 8.8 Hz, 2H, H-2′,H-6′), 7.82 (d, J = 8.4 Hz, 2H, H-3′, H-5′); 13C NMR (100 MHz, CD3OD) δ 40.1, 51.4, 115.1, 117.9, 124.9, 127.0, 127.7, 132.4, 138.4, 140.5, 165.5. HRESIMS (positive mode) m/z 263.1181 [M + H]+ (calcd for C17H14N2O, 263.3021),
N-(2,3-dihydro-1H-inden-2-yl)-cinnamamide (B10) (0.3061 g, 93.75% yield, black solid) (Fig. 3), IR (KBr) vmax: 3,269, 1,655, 1,566, 1,360, 1,238, 974 cm-1; 1H NMR (400 MHz) δ 2.88 (dd, J = 16, 4.4 Hz, 2H, H-2), 3.37 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 4.88 (m, 1H, H-1), 6.31 (d, J = 15.6 Hz, 1H, H-3′), 7.19 (dd, J = 6.2, 2.8 Hz, 2H, H-7, H-8), 7.25 (dd, J = 8.6, 2.0 Hz, 2H, H-6, H-9), 7.34 (dd, J = 6.8, 1.6 Hz, 3H, H-5′, H-9′, H-7′), 7.47 (dd, J = 7.6, 4.0 Hz, 2H, H-6′,H-8′), 7.62 (d, J = 15.6 Hz, 1H, H-2′); 13C NMR (100 MHz, CD3OD) δ 40.2, 50.9, 120.6, 124.9, 126.8, 127.8, 128.8, 129.7, 134.8, 140.9, 141.1, 165.6. HRESIMS (positive mode) m/z 264.1387 [M + H]+ (calcd for C18H17NO, 264.3296)
N-(2,3-dihydro-1H-inden-2-yl)-3,4-dimethoxybenzamide (B11) (0.3725 g, 70.83% yield, white solid) (Fig. 3), IR (KBr) vmax: 3,295, 1,627, 1,543, 1,271, 1,235, 1,134 cm-1; 1H NMR (400 MHz) δ 2.93 (dd, J = 16.4, 4.8 Hz, 2H, H-2), 3.43 (dd, J = 16.4, 7.2 Hz, 2H, H-3), 3.90 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 4.91–4.97 (m, 1H, H-1), 6.81 (d, J = 8.4 Hz, 1H, H-6′), 7.18 (d, J = 8.4 Hz, 2H, H-6, H-9), 7.19 (d, J = 8.4 Hz, 2H, H-7, H-8), 7.26 (t, J = 7.4 Hz, 1H, H-5′), 7.41 (d, J = 7.2 Hz, 1H, H-4′); 13C NMR (100 MHz, CD3OD) δ 40.3, 51.1, 56.0, 110.2, 110.8, 119.2, 124.9, 126.9, 127.3, 140.9, 149.1, 151.8, 166.9. HRESIMS (positive mode) m/z 298.1445 [M + H]+ (calcd for C18H19NO3, 298.3393).
Biological evaluation
Cytotoxicity screening assay (MTS Assay)
HepG2 cells were seeded onto 96-well plates at a density of approximately 8,000 cells/well and grown at 37°C in a humidified incubator supplemented with 5% (v/v) CO2 for 24 hours. Subsequently, the cells were treated with the concentrations of compounds ranged from 3.13 to 50 μM. The negative and positive controls were treated with dimethylsulfoxide (DMSO) and vincristine sulfate, respectively, where both are known as standard drugs in the treatment of liver cancer (Nissen and Wolski, 2007). The plate was then incubated for 72 hours at 37°C in 5% (v/v) CO2 incubator. After 72 hours of incubation, the culture was assayed using CellTiter 96 Aqueous One Solution Cell Proliferation Assay System (Promega). Approximately 20 μl of 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) solution was added into each well and incubated for 3.5 hours in the humidified 5% (v/v) CO2 incubator at 37°C. The wells with complete medium and MTS solution without cells were used as blanks. The absorbance was determined at 490 nm using Glomax-Multi Detection System (Promega).
Promoter analysis assay (Dual-Glo luciferase assay)
Plasmid extraction was conducted using Qiagen Midi Kit (Qiagen, Germany) according to the manufacturer’s protocols. The plasmids contributed by Yonsei University, Korea, were preserved in glycerol stock. The culturing method for bacteria was used during the experiment since the plasmids were located within the bacteria. Before transfection, HepG2 cells were seeded onto 96-well plates at a density of approximately 40,000 cells/well and further optimized with 100 μl of cells in media. After 24 hours, the cells were cultured in serum-reduced medium OPTIMEM and the transient transfection was carried out using Lipofectamine LTX and PLUS reagents (Invitrogen, USA) according to the manufacturer’s recommendation. The cells were transiently transfected with pGL3 plasmid harboring firefly luciferase reporter gene. As an internal control for transfection efficiency, the cells were cotransfected with a Renilla luciferase-encoding reference plasmid, pRL-tk. After 5 hours of incubation, the transfected cells were treated with compounds at concentrations ranged from 3.13 to 50 μM. The two-fold serial dilution technique was used to prepare the working solutions. The negative control was treated with DMSO, whereas the positive control was treated with berberine sulfate with the optimized concentration of 20 μM. The treated cells were incubated for 24 hours at 37°C in a humidified incubator supplemented with 5% (v/v) CO2. Dual-Glo Luciferase Assay System was carried out according to the manufacturer’s instructions to identify the compound activities toward the expression of pGL-3 proprotein convertase subtilisin/kexin type 9 (PCSK9) promoter. About 90 μl of Dual-Glo® Luciferase Reagent purchased from Promega (USA) was added to each well. The plate was later incubated for 10 minutes in a biohazard safety cabinet type II and measured with Glomax-Multi Detection System. After that, 90 μl of Dual-Glo® Stop and Glo® Reagent purchased from Promega (USA) was added to each well and incubated for 10 minutes. The plate was once again measured with Glomax Multidetection System for luminescence reading. The data collection was generated by measuring the firefly luciferase and Renilla luciferase activities. The results from the measurement of firefly luciferase were divided with the results from Renilla luciferase. The calculations were normalized against the negative control.
RESULTS AND DISCUSSION
Synthesis of aaptaminoids
Aaptaminoids are benzo[de][1,6]naphthyridine alkaloid compounds, found abundantly in marine sponges from the genera of Aaptos. Even though they display a wide range of chemical and biological importance (Larghi et al., 2014; Rashid et al., 2018), the discovery of the biological activities of compound related to CVD is still scarce. In the previous work, several aaptaminoids such as aaptamine, 9-methoxyaaptamine, 4-N-methylaaptamine, and 9-methoxyaaptamine were found to increase the expression of both SRB1 and PPARγ genes, which are the therapeutic target for atherosclerosis treatment (Habsah et al., 2017). The total synthesis and chemical structure modification of aaptamine through organic synthesis has been reviewed by Larghi et al. (2014). We prepared N-benzyl derivative of aaptamine which is N1,N4-bisbenzylaaptamine from its parent compound (aaptamine) through benzylation at N1,N4 using K2CO3 as base and dry DMF as solvent (73% yield). The synthesis of N4-[(3,4,5-trimethoxy)benzyl]aaptamine (31% yield) and N1-[(3,4,5-trimethoxy)benzyl]aaptamine (25% yield) was prepared using KH as base and dry DMF as solvent. The same compound was also synthesized by Pettit et al. (2004) together with several other N-benzyl derivatives of aaptamines. The studies on N-alkylation were also reported by Amin et al. (2018) and Abdul Rashid et al. (2014). Their work provides information on compounds that were cytotoxic to Acanthamoeba castellani and displayed antibacterial activity.
Synthesis of benzamide–aminoindane derivatives
Methyl benzoates were previously isolated from A. planci (Mat Lazim et al., 2016). To synthesize its derivatives, we used benzamides as the starting material. In this work, the synthesis of the benzamide–aminoindane derivatives was carried out to study the potential of the compounds as PCSK9 inhibitors. Benzamide compounds are reported to have cholesterol-lowering property. According to Drazic et al. (2015), novel amide amino-beta-lactam derivative was capable to reduce the cholesterol level up to 53% through in vivo study. Furthermore, a series of novel substituted benzamides analogs comprised azaspiro rings were able to induce the transcription of the Apo A-I gene. It was reported that the trifluoromethyl-substituted benzamide containing an azaspiro ring is a promising backbone for designing Apo A-I transcriptional upregulator and could be a viable lead for the development of new drugs to prevent and treat atherosclerosis in the future (Du et al., 2012). In addition, several patents claimed that aminoindane derivatives could serve as cholesterol-lowering substances (Follmann et al., 2013; Kathawala, 1980; Wohlfart et al., 2004). All these successful studies stand as a basis on why we pursue to synthesize benzamide–aminoindane to study its potential as PCSK9 inhibitors. In this study, eleven benzamide–aminoindane (B1–B11) compounds were successfully synthesized with percentage yield ranging from 33.38% to 99.12%. Compounds B2, B3, B9, and B11 were new compounds, whereas the other derivatives are already known.
Biological evaluation
Cytotoxic activity against HepG2 cell line
Compounds A1–A3 and B1–B6 were tested for cytotoxicity activity against hepatocellular carcinoma liver cancer cell (HepG2) using MTS assay. This method is a colorimetric assay that is based on the conversion of tetrazolium salt into a colored formazan by the metabolic activity of the living cells (Aslanturk, 2017). The amount of produced formazan depends on the viable cell number in the culture (Aslanturk, 2017). Cytotoxicity test plays an important role in evaluating the hazardous effects and anticancer properties. These characteristics can be induced by testing the compounds and predicting the in vivo human toxicity. In drug discovery, compound screening assays for hit discovery are commonly run at 1–10 μM (Hughes et al., 2011) with total therapeutic plasma concentrations for most marketed drugs which are below 10 μM (Williams et al., 2004). This implies that compounds that have IC50 ≤ 10 μM are considered toxic to the cells.
In Figure 4, aaptaminods (A1, A2, and A3) demonstrated no 50% of cell growth inhibition occurred at a concentration less than 10 μM (IC50 > 10 μM), which indicates that the studied compounds (aaptamine derivatives) were nontoxic toward HepG2 cells. The half-maximal inhibitions were only exhibited by compounds A1 and A2 at the highest concentration (50 μM). As for methyl benzoate derivatives, Figure 5 shows that there is no 50% inhibition of cell growth occurred when treated with compounds B1, B2, B3, B5, and B6 at any concentrations. On the contrary, compound B4 inhibited 50% of cell growth but only at the highest concentration of 50 μM. Thus, it is concluded that all methyl benzoate derivatives synthesized in this work were nontoxic against HepG2 cell line. These compounds were further evaluated with PCSK9 transcriptional promoter activity.
PCSK9 inhibitory activity
Several nutraceuticals have been reported to act as a potential treatment for atherosclerosis in preclinical and clinical studies which consist of omega-6-polyunsaturated fatty acids, hydroxytyrosol, allicin, berberine, phytosterols, lycopene, flavonols including epicatechin and catcechin, Vitamins C and E, carnosine, coenzyme Q10, curcumin, lycopene, and resveratrol (Moss and Ramji, 2016; Moss et al., 2018). Among them, several nutraceuticals such as berberine, curcumin, polydatin (resveratrol-3-O-b-mono-d-glycoside), Xuezhikang and red yeast rice, omega-3-fatty acid, quercetin-3-O-b-d-glucoside, tanshinone IIA, sauchinone, and Phaleria macrocarpa fruit extract were found to inhibit the expression of PCSK9 gene (Chae et al., 2018; Chen et al., 2016; Cicero and Ertek, 2009; Momtazi et al., 2017). A total of 1041 compounds from the National Institute of Neurological Disorders and Stroke library were screened for PCSK9 modulatory activity using in vitro cell-based LDL uptake model, and it was found that colchicine, a Food and Drugs Association (FDA)-approved drugs for the treatment of gout, inhibits the PCSK9 degradation of LDLR (Xu and Liu, 2013). Besides screening the inhibition of PCSK9 gene expression in vitro, several in silico screenings in inhibiting the PCSK9 protein of thousands of small molecules available from the established chemical libraries were also done (Lammi et al., 2016; Min et al., 2015; Reddy et al., 2016; Taechalertpaisarn et al., 2018). Two compounds, ZINC85625485 and ZINC85625406, which possess an ability to act as potential inhibitors for PCSK9, are executed using the screening of zinc database, a technique among several of the in silico screening methods. (R)-6-Amino-2-((S)-2-(4-((R)-2-((R)-4-(2-amino-2-oxoethyl)-2,5-dioxoimidazolidin-1-yl)-3-mercaptopropanoyl)piperazin-1-yl)-4-methylpentanamido)-N-((S)-2-oxotetrahydrofuran-3-yl)hexanamide, (S)-2-(4-((R)-2-((R)-4-(2-amino-2-oxoethyl)-2,5-dioxoimidazolidin-1-yl)-3-mercaptopropanoyl)piperazin-1-yl)-N-((R)-5-guanidino-1-oxo-1-(((S)-2-oxotetrahydrofuran-3-yl)amino)pentan-2-yl)-4-methylpentanamide, and their corresponding analogs were small molecules that can serve as potential inhibitors of the PCSK9–LDLR interaction (Taechalertpaisarn et al., 2018). The modified crystal structure of PCSK9 from protein databank, CB_36 (#7926604), and its analog was found to be effective in silico from out of top 100 chemicals with the highest docking score from the ChemBridge Express collection dock. Interestingly, CB_36 is also effective in the in vitro and in vivo models of LDL-C uptake. Thus, both natural and synthetic compounds can serve as potential hits and lead in the exploration of finding new potential PCSK9 inhibitor.
 | Figure 4: Cytotoxic activity of aaptaminoids against HEPG2 cell line N4- Bisbenzylaaptamine [A1]; N4-[3,4,5-Trimethoxyl)benzyl]aaptamine [A2]; N1- [3,4,5–Trimethoxyl)benzyl]aaptamine [A3]. [Click here to view] |
PCSK9 promoter transcriptional activity in the analysis was carried out using Dual-Glo Luciferase Assay System (Promega) to elucidate the potential of isolated compound aaptamine, methyl benzoate, and their derivatives in reducing the promoter activity of PCSK9 gene. To study the effects of selected compounds on the PCSK9 promoter transcriptional activity, HepG2 cells were transfected with pGL3 plasmid harboring firefly luciferase reporter gene and cotransfected with a Renilla luciferase-encoding reference plasmid, pRL-tk, for transfection efficiency. The same concentration was used for cytotoxicity assay except for compounds A1 and A2 (highest concentration: 25μM). The cells treated with berberine sulfate were used as the positive control because berberine can suppress PCSK9 expression in cholesterol metabolism (Dong et al., 2015).
The luciferase activity produced by the treated samples was compared to that of the untreated control, assigned as 1. If the number of the fold change was less than the control (<1), the compound was considered to have a positive activity. On the contrary, any compound that showed the number of fold change of more or equal than 1 (≥1) was considered to have a negative activity and described as nonactive.
 | Figure 5: Cytotoxic activity of benzamides against HEPG2 cell line. B1: N-(2,3-Dihydro-lH-inden-2-yl)benzamide, B2: N-(2,3-dihydro-lH-inden-2-yl)-2-methoxybenzamide, B3: N-(2,3-dihydro-lH-inden-2-yl)-3-methoxybenzamide, B4: N-(2,3- dihydro-lH-inden-2-yl)-4-methoxybenzamide, B5: N-(2,3-dihydro-lH-inden-2-yl)-4-fluorobenzamide, B6: N-(2,3-dihydrolH- inden-2-yl)-4-chlorobenzamide. [Click here to view] |
Figure 6 shows the PCSK9 inhibitory activity of aaptaminoids (A and A1–A3). All aaptaminoids A1–A3 possessed a potential ability to inhibit the transcriptional activity of PCSK9 promoter. Compounds A1 (fold change = 0.35 at 6.25 μM) and A3 (fold change = 0.49 at 3.125 μM) showed the highest inhibition for transcriptional activity of PCSK9 promoter, followed by compounds A2 (fold change = 0.58 at 12.5 μM) and A (fold change = 0.61 at 50 μM). Compound A was found to reduce the promoter activity in a dose-dependent manner. In general, aaptamine and its derivatives here gave comparable findings in the inhibition of the transcriptional activity of PCSK9 with berberine sulfate. Further studies on the underlying mechanism on how compound A inhibits the transcriptional activity of PCSK9 promoter are still ongoing.
Meanwhile, Figure 7 shows the PCSK9 transcriptional promoter activity of benzamide–indane derivatives (B1–B5). Among the five compounds tested, only compound B2 showed the reduction of PCSK9 transcriptional promoter activity. Compound B2 showed a dose-dependent manner in reducing the PCSK9 promoter activity starting from 6.25 to 50 μM.
Overall, the results from this PCSK9 transcriptional promoter activity screening suggest that aaptaminoids (A and A1–A3) and benzamide derivative (B2) possessed the ability to be developed as PCSK9 inhibitor in reducing the progression of atherosclerosis. In the case of aaptaminoids, all derivatives (A1–A3) displayed higher PCSK9 inhibition activity than its parent molecule aaptamine. Compounds B2–B4 only differed in the position of the methoxy group in the benzene ring (Figure 4), and only B2 with the position of methoxy group and C-2′ next to the carbonyl group can inhibit the expression of PCSK9 gene. The presence of fluoride functional group at C-4′ did not contribute in inhibiting the PCSK9 activity. These compounds can be further analyzed for their potential role in the treatment of atherosclerosis as drug candidates.
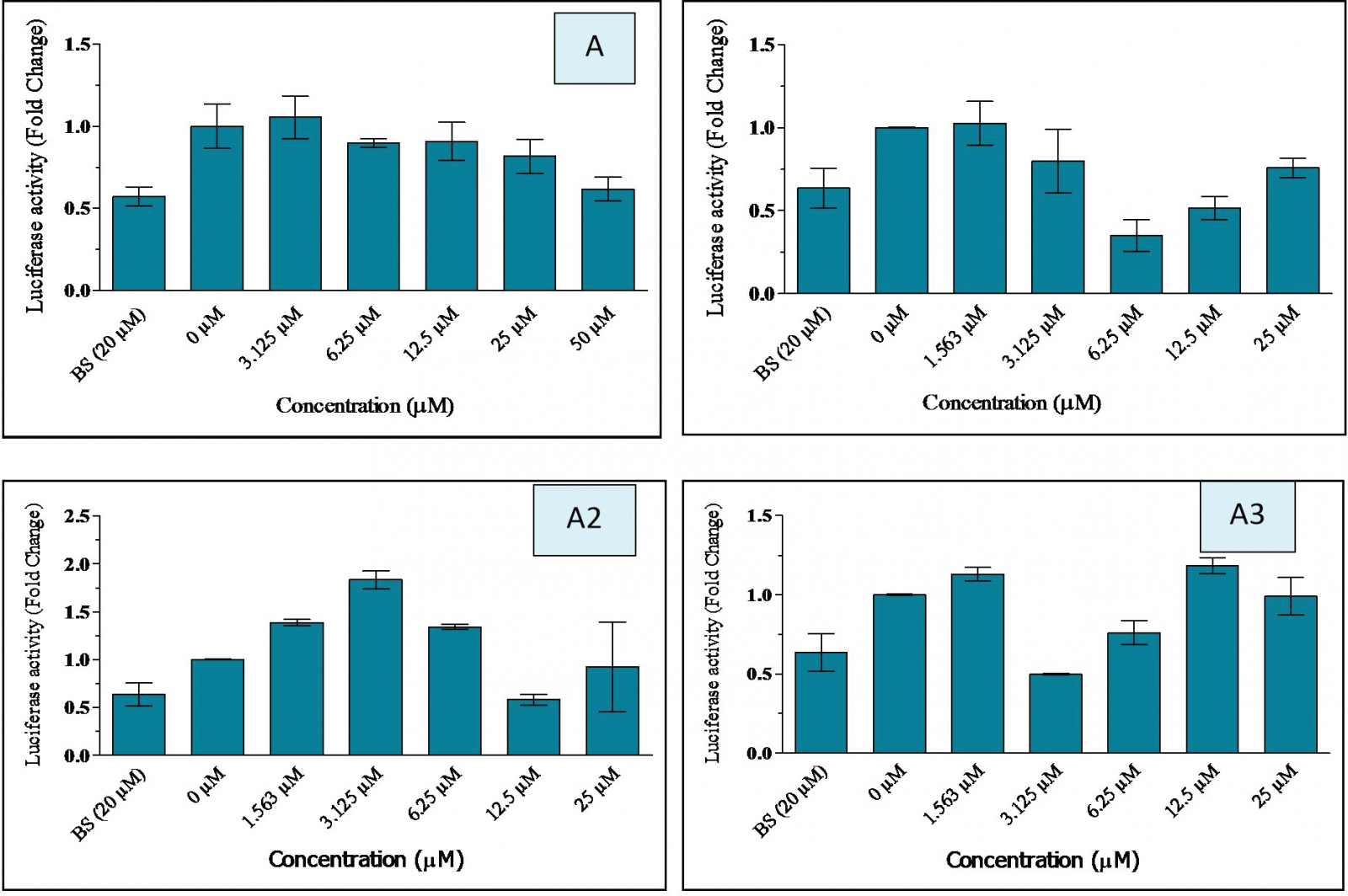 | Figure 6: Inhibitory activity of aaptamine and its derivatives on PCSK9 gene measured by luciferase assay A: Aaptamine, A1: N1,N4-Bisbenzylaaptamine, A2: N4- [(3,4,5-trimethoxyl)benzyl]aaptamine, A3: N1-[3,4,5-trimethoxyl)benzyl]aaptamine, BS: berberine sulfate, HepG2 cells were cotransfected with 2 μg pGL3-PCSK9 (D1) and 0.5 μg pRL-TK. [Click here to view] |
.png) | Figure 7: Inhibitory activity of methyl benzoate derivatives on PCSK9 gene measured by luciferase assay B1: N-(2,3-dihydro-lH-inden-2-yl)benzamide, B2: N-(2,3- dihydro-lH-inden-2-yl)-2-methoxybenzamide, B3: N-(2,3-dihydro-lH-inden-2-yl)-3-methoxybenzamide, B4: N-(2,3-dihydro-lH-inden-2-yl)-4-methoxybenzamide, B5: N-(2,3-dihydro-lH-inden-2-yl)-4-fluorobenzamide, BS: berberine sulfate, HepG2 cells were cotransfected with 2 μg pGL3-PCSK9 (D1) and 0.5 μg pRL-TK. [Click here to view] |
CONCLUSION
In this work, three known aaptamine derivatives were synthesized: N1,N4-bisbenzylaaptamine, N4-[(3,4,5-trimethoxyl)benzyl]aaptamine, and N1-[(3,4,5-trimethoxyl)benzyl]aaptamine. All three compounds have the potential to downregulate the PCSK9 gene expression. Of eleven synthesized methyl benzoate derivatives, five of them were evaluated for the possibility to inhibit the PCSK9 gene expression. We found only one, which is N-(2,3-dihydro-1H-inden-2-yl)-2-methoxybenzamide could inhibit the expression of the PCSK9 gene. Thus, these compounds can be used as a hit or lead compound for an alternative drug in reducing the burden of atherosclerosis disease.
ACKNOWLEDGMENT
This study was funded by the Ministry of Higher Education, Malaysia, under the Transdisciplinary Research Grant Scheme (TRGS) vote 59422.
CONFLICT OF INTEREST
Authors declare that they do not have any conflicts of interest.
REFERENCES
Abdul Rashid FNA, Asari A, Habsah M, Mohd Nor SM. Synthesis and antibacterial study of aaptamine derivatives. Asian J Chem, 2014; 26(20):6903–7. CrossRef
Abdul Razak MF, Asari A, Hamzah AS, Addis SNK, Habsah M. Synthesis, characterisation and antibacterial activity of Hystatin 2 derivatives. J Chem Pharm Res, 2015; 7(4):330–837.
Aiman U, Najmi A, Khan RA. Statin induced diabetes and its clinical implications. J Pharmacol Pharmacother, 2014; 5(3):181–5. CrossRef
Amin NM, Hamdin MS, Azarnudin MAT, Asari A, Abdul Rashid FNA, Mohd Nor SM. Cytotoxic effect of aaptamine and its derivatives on Acanthamoeba castellani (IMR isolates). Sci Int (Lahore), 2018; 30(2):309–13. and disadvantages. In: Larramendy M, Solonesky S. (eds.). Genotoxicity: a predictable risk to our actual world, Intech Open, London, UK, pp 4–5, 2017.
Aslanturk OS. In vitro cytotoxicity and cell viability assays: principles, advantages
Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanism of plaque formation and rupture. Circulation Res, 2014; 114:1852–66. CrossRef
Blunt JW, Copp BR, Keyzers RA, Munro, MHG, Prinsep MR. Marine natural products. Nat Prod Rep, 2015; 32:116–211. CrossRef
Bowling JJ, Pennaka HK, Ivey K, Wahyuono S, Kelly M, Schinazi RF, Valeriote FA, Graves DE, Hamann MT. Antiviral and anticancer optimization studies of the DNA-binding marine natural product aaptamine. Chem Biol Drug Des, 2008; 71(3):205–15. CrossRef
Cariou B, Le May C, Costet P. Clinical aspects of PCSK9. Atherosclerosis, 2011; 216:258–65. CrossRef
Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis, 2013; 228:18–28. CrossRef
Chae HS, You BH, Kim DY, Lee H, Ko HW, Ko HJ, Choi YH, Choi SS, Chin YW. Sauchinone controls hepatic cholesterolhomeostasiss by negative regulation of PCSK9 transcriptional network. Sci Rep, 2018; 8(1):6737; doi:1010381541598-018-24935-6 CrossRef
Chen HC, Chen PY, Wu MJ, Tai MH, Yen JH. Tanshinone IIA modulates low density lipoprotein uptake via down-regulation of PCSK9 gene expression in HepG2 cells. PLoS One, 2016; 11(19):1–18; doi:10.1371/journal.pone.0162414 CrossRef
Cicero A, Ertek S. Metabolic and cardiovascular effects of berberine:from preclinical evidences toclinical trial results. Clin Lipidol, 2009; 4(5):553–63. CrossRef
Dong B, Li H, Singh AB, Cao A, Liu J. Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNFI alpha protein expression through ubiquitin- proteasome degradation pathway. J Biol Chem, 2015; 290(42):4047–58. CrossRef
Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSKi9 gene expression through HNF1alpha and SREBP2: mechanisms for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res, 2010; 51:1486–95. CrossRef
Drazic T, Sachdev V, Leopold C, Patankar JV, Malnar M, Hecimovic S, Franks SL, Habus I, Kratky D. Synthesis and evaluation of novel amide amino-β-lactam derivatives as cholesterol absorption inhibitors. Bioorg Med Chem, 2015; 23(10):2353–9. CrossRef
Du Y, Yang Y, Jiang W, Wong L, Jia XJ, Si SY, Chen XF, Hong B. Substituted benzamides containing azaspiro rings as upregulators of apolipoprotein A-1 transcription. Molecules, 2012; 17:7379–86. CrossRef
Eguchi K, Fujiwara Y, Hayashida A, Horlad H, Kato H, Rotinsulu H, Losung F, Mangindaan REP, Voogd NJD, Takeya M, Tsukamoto S. Manzamine A, a marine-derived alkaloid, inhibits accumulation of cholesterol ester in macrophages and suppresses hyperlipidemia and atherosclerosis in vivo. Bioorg Med Chem, 2013; 21:3831–8. CrossRef
Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Comput Sci, 2003; 43(1):218–27. CrossRef
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J, Nochur SV, Kotelianski V, Horton J, Mant T, Chiesa J, Ritter J, Munisamy M, Vaishnaw AK, Gollob JA and Simon A. Effect of a RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type9 (PCSK9) and the concentration of serum LDL cholesterolin healthy volunteers: arandomised, single-blind, placebo-controlled, phase1trial. Lancet, 2014; 383:60–8. CrossRef
Follmann M, Hatchtel S, Hessler G, Kleeman HW, Maier T, Cort GMC, Struebing G, Thiers B, Wong LH. Derivatives of aminoindanes, their application in therapeutics, United States Patent US 2013/0131034 A1.
Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol, 2009; 27:165–97. CrossRef
Galm U, Shen B. Natural product drug discovery: the times have never been better. Chem Biol, 2007; 14:1098–104. CrossRef
Go A, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics—2014 update a report: from the American Heart Association. Circulation, 2014; 129:399–410. CrossRef
Goldstein JL, Brown MS. The LDL receptor. Atherioscler. Thromb Vasc Biol, 2009; 29:431–8. CrossRef
Grabowski K, Schneider G. Properties and architecture of drugs and natural products revisited. Curr Chem Biol, 2007; 1:115–27. CrossRef
Habsah M, Rosmiati, Muhammad TSTM, Andriani Y, Bakar K, Ismail N, Saidin J, Latip J, Musa N, Parerengi A. Potential secondary metabolites from marine sponge Aaptos aaptos for atherosclerosis and vibriosis treatments. Nat Prod Comm, 2017; 12(8):1227–30. CrossRef
Hu Y, Chen J, Hu G, Yu J, Zhu X, Lin Y, Chen S, Yuan J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar Drugs, 2015; 13:202–21; doi:10.3390/md13010202 CrossRef
Hughes JP, Rees S, Kalindijian SB, Philpott KL. Principles of drug discovery. British J Pharmacol, 2011;162:1239–49. CrossRef
Jeong HJ, Lee HS, Kim KS, Yoon D, Park SW. Sterol-dependent regulationof proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res, 2008; 49:390–409. CrossRef
Jia YJ, Xu RX, Sun J, Tang Y, Li JJ. Enhanced circulating PCSK9 concentration by berberine through SREBP-2 pathway in high fat diet-fed rats. J Translation Med, 2014; 12:103–10. CrossRef
Kathawala FG, Benzocycloalkylamides, United Stes Patent. 1980; 4(185):118.
Koren MJ, Lundovist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H. Anti-PCSK9 monotherapy for hypocholesterolemia: the MENDEL-2 randomozed, control phase III clinical trial of evolucumab. J Am Col Cardiol, 2014; 63(23):2532–40. CrossRef
Lambert G, Sjouke B, Choque B, Kastelein JJ and Hovingh GK. ThePCSK9 decade. J Lipid Res, 2012; 53:2515–2524. CrossRef
Lammi C, Zanoni C, Aiello G, Arnoldi A, Grasioso G. Lupin peptides modulate the protein-protein interaction of PCSK9 with the low density lipoprotein receptor in HepG2 cells. Sci Rep, 2016;6:29931–43; doi:10.1038/srep29931 CrossRef
Larghi EL, Bohn ML, Kaufman TS. Aaptamine and related products. Their isolation, chemical syntheses, and biological activity. Tetrahedron, 2014; 65:4257–82. CrossRef
Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclearfactor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem, 2009; 284:28885–95. CrossRef
Lindholm MW, Elmén J, Fisker N, Hansen HF, Persson R, Møller MR, Rosenbohm C, Ørum H, Straarup EM, Koch T. PCSK9 LNA antisense oligonucleotides inducesustained reduction of LDL cholesterol in nonhuman primates. Mol Ther, 2012; 20:376–81. CrossRef
Luo Y, Coob RE, Zhao H. Recent advances in natural product discovery. Curr Opinion Biotechnol, 2014; 30:230–7. CrossRef
Maiolino G, Rossitto G, Caielli P, Bisogni V, Rossi GP, Calo LA. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm, 2013; 2013:714653. CrossRef
Mat Lazim NH, Asari A, Mohamad F, Muhammad TST, Ismail N, Ahmad A, Taib M, Habsah M. Potential anti-atherosclerotic compound isolated from Acansther planci. Res J Pharm Biol Chem Sci, 2016; 7(1):482–7.
Mayer AMS, Lehmann VKB. Marine pharmacology in 1998: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, anthelminitic, antiplatelet, antiprotozoal, and antiviral activities with actions on the cardiovascular, endocrine, immune, and nervous systems, and other miscellaneous mechanisms of action. Pharmacologist, 2000; 42:62–9.
Min DK, Lee HS, Lee N, Lee CJ. Insilico screening of chemical libraries to develop inhibitors that hamper the interaction of PCSK9 with the LDL receptor. Yonsei Med J, 2015; 56(5):1251–7. CrossRef
Momtazi AA, Banach M, Pirro M, Katsiki N, Sahebkar A. Regulations of PCSK9 by nutraceutical. Pharmacol Res, 2017; 120:157–69. CrossRef
Moss JWE, Ramji DP. Nutraceutical therapies for artherosclerosis. Nat Rev Cardiol, 2016; 13(9):513–32; doi:10.1038/nrcardio.2016.103 CrossRef
Moss JWE, Williams JO, Ramji DP. Nutraceutical as therapeutic agents for atherosclerosis. Biochim Biophys Acta Mol Basis Dis, 2018; 1864:1562–72. CrossRef
Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat P rod, 2007; 70(3):461–77. CrossRef
Nissen SE, Wolski K. Effects of rosiglitazone on the risk of myocardial infarction and death from cardiovascular diseases. New England J Med, 2007; 356(24):2457–71. CrossRef
Pettit GR, Hoffmann H, Herald DL, Blumberg PM, Hamel E, Schmidt JM, Chang Y, Pettit RK, Lewin NE, Pearce LV. Antineoplastic agents. 499. Synthesis of hystatin 2 and related 1H-benzo[de][1,6]-naphthyridinium salts from aaptamine. J Med Chem, 2004; 47:1775–82. CrossRef
Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A andSeidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem, 2009; 284:28856–64. CrossRef
Proksch PR, Edrada A, Ebel R. Drugs from the seas—current status and microbiological implications. Appl Microbiol Biotechnol, 2002; 59:25–34.
Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA andHorton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc Natl Acad Sci USA, 2005; 102:5374–9. CrossRef
Rashid ZM, Ali AM, Douzenel P, Bourgougnon N, Shaari K, Andriani Y, Muhammad TST, Habsah M. Phenolics, fatty acids composition and biological activities of various extracts and fractions of Malaysian Aaptos aaptos. Asian Pacific J Trop Biomed, 2018; 8(11):554–64. CrossRef
Reddy VV, Reddy MCH, Suganya RP. Identification of potential inhibitors for lowering cholesterol level by inhibiting proprotein convertase sutilisin kexin 9. Asian J Pharm Res, 2016; 9(6):230–4. CrossRef
Richard-Deichmann MD, Carl-Lavie MD, Samuel-Andrew MD. Coenzyme Q10 and statin-induced mitochondrial dysfunction. Ocshner J, 2010; 10:16–21.
Roth EM, Taskinem MR, Ginsberg HN, Kaestelein JJ, Colhaun HM, Robinson JG, Merlet L, Pordy R, Baccara-Dinet MT. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetemibe in patients with hypercholesterolemia: results in a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol, 2014; 176(1):55–61. CrossRef
Rubinstein A, Izkhakov E. Statins: an effective anti-atherosclerosis therapy. Isr Med Assoc J, 2002; 4:456–7.
Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res, 2014; 114:1022–36. CrossRef
Steinberg D, Witztum JL. History of discovery: oxidised low-densitiy lipoprotein and atheroclerosis. Arteriosclerosis Thromb Vasc Biol, 2010; 30:2311–6. CrossRef
Stock J. Highlights of the 83rd European Atherosclerosis Society (EAS) annual Congress, Glasgow 22-25 March. Atherosclerosis, 2015; 242:45–7. CrossRef
Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol, 2015; 209:13–22. CrossRef
Taechalertpaisarn J, Zhao B, Liang X, Burgess K. Small molecule inhibitors of the PCSK9-LDLR interaction. J Am Chem Soc, 2018; 140:3242–9. CrossRef
Tomkin GH, Owens D. LDL as a cause of atherosclerosis. Open Atheroscler Thromb J, 2012; 5:13–21. CrossRef
Verbeek R, Stoekenbroek RM, Hovingh GK. PCSK9 inhibitors: novel therapeutic agents for the treatment of hypercholesterolemia. Eur J Pharmacol, 2015; 763(Pt A):38–47; doi:10.1016/j.ejphar.2015.03.099i CrossRef
Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug-drug interactions for UDP-glucuronosyl transferase substrates: a pharmacokinetics explanation for typically observed low exposure (AUCI/AUC) ratios. Drug Metabol and Disposit, 2004; 32(11):1201-1208. CrossRef
Wohlfart P, Suzuki T, Dharanipragada RM, Safarova A, Walser A, Strobel H, 2004. United States Patent US 6,812,253 B2.
Xia X, Li Y, Su Q, Huang Z, Shen Y, Li W, Yu C. Inhibitory effects of Mycoepoxydiene on macrophage foam cell formation and atherosclerosis in ApoE-deficient mice. Cell Biosci, 2015; 5:23. CrossRef
Xu J, Murphy SL, Kochanek KD, Arias E. Mortality in the United States, 2015. NCHS Data Brief , No. 267. December 2016. https://www.cdc.gov/nchs/data/databriefs/db267.pdf
Xu W, Liu L. An invitro cell-based LDL uptake model for screening PCSK9 modulators. Bioequival Bioavailabil, 2013; 5(7):248–52.
Zhou X, Xu T, Yang X-W, Huang R, Yang B, Tang L, Liu Y. Chemical and biological aspects of marine sponges of the Genus Xestospongia. Chem Biodivers, 2010; 7:2201–27. CrossRef
Zhu M, Gao H, Wu C, Zhu T, Che Q, Gu Q, Guo P, Li D. Lipid-lowering polyketides from a soft coral-derived fungus Cladosporium sp. TZP29. Bioorg Med Chem Lett, 2015; 25:3606–9. CrossRef