
INTRODUCTION
Currently, the gradual increase of infections caused by the higher resistance of bacteria and fungi and the widespread use of antibiotics are still the major concerns of human illness or even death due to the dramatically reduced effectiveness of drugs. To overcome the microbial resistance and their emergence, developing a new alternative of the potent antibacterial and antifungal agents with novel scaffolds is essential (Ghannay et al., 2017, 2020a). The α-glucosidase plays a vital role in carbohydrate metabolism and glycoprotein biosynthesis. Its inhibition by acarbose, miglitol, or voglibose is one of the important therapeutic approaches for the management of degenerative diseases, such as type 2 diabetes mellitus and also used to treat anti-HIV (Rawlings et al., 2009; Vichayanrat et al., 2020), anticancer (Pili et al., 1995), and anti-hepatitis diseases (Zitzmann et al., 1999). These inhibitors were associated with side effects, such as diarrhea and abdominal discomfort and possess the weaker IC50 values (Chougale et al., 2009). Therefore, there is an urgency to discover the safe and effective inhibitors of this key enzyme for the control of diabetic disorders.
In this context, we find that many families are eligible for these activities such as isoxazolidines (Chiacchio et al., 2016; Ghabi et al., 2020), α-amino-lactones (Abda et al., 2014), and amino acids and their derivatives (Zhou et al., 2019). The research work of our team revolves around the synthesis of new series of enantiopure polyfunctional isoxazolidines (Abda et al., 2016; Aouadi et al., 2006, 2013; Brahmi et al., 2016b), which are in turn transformed into aminoalcohol and/or amino acid derivatives (Aouadi et al., 2007, 2008, 2012b), such as trifluoromethylated isoxazolidines (Ghannay et al., 2020a), 4-hydroxyisoleucine (Aouadi et al., 2007, 2012b), and its analogs (Aouadi et al., 2008, 2009). We can also give as an example for the synthesis of 4-hydroxy-L-ornithine (Aouadi et al., 2012a), 4-hydroxyproline derivatives (Brahmi et al., 2016a), 4-hydroxypyrrolidine derivatives (Cecioni et al., 2015), and pyrrolidine-2,5-dione derivatives (Ghannay et al., 2019).
In continuation of the drug discovery research toward the synthesis of bioactive heterocyclic compounds on potential antimicrobial and antidiabetic agents (Ghannay et al., 2017, 2020a, 2020b), we herein report the biological evaluation of antimicrobial and α-glucosidase inhibitory properties of some enantiopure cycloalkylglycines derivatives. Furthermore, the structure–activity relationship was thoroughly discussed. In addition, in order to get insights into their pharmacodynamic and pharmacokinetic profiles, the compounds were subjected to in silico molecular property and absorption, distribution, metabolism, and excretion (ADME) predictions using Molinspiration and SwissADME online software, respectively.
MATERIALS AND METHODS
Chemistry
The various compounds 1–15 were synthesized based on the cycloaddition reaction of a chiral nitrone with various cycloalkenes as was reported in the previous work (Abda et al., 2014) (Scheme 1). Basic processing of α-amino-γ-lactones 1–4 has provided the desired α-amino acids 11–15 (Abda et al., 2014). The structures of compounds 1–15 have been unambiguously reported (Abda et al., 2014).
Computational study
The molecular profile and druglikeness of the tested compounds have been assessed using Molinspiration software (https://www.molinspiration.com/cgi-bin/properties) (Ghannay et al., 2017) by calculating the different parameters as shown in Table 3.
The pharmacokinetic and druglikeness properties of the synthesized drugs, such as passive human gastrointestinal absorption (GI), blood–brain barrier (BBB) permeation, skin penetration coefficient, substrate or non-substrate of the permeability glycoprotein (P-gp), and interaction of molecules with five major isoforms of the human cytochromes P450 interfering in the metabolism of numerous endogenous and exogenous compounds, have been predicted using SwissADME online server (http://www.swissadme.ch/) (Daina et al., 2017). Furthermore, the medicinal chemistry characteristics such as PAINS for pan-assay interference compounds or promiscuous compounds, Brenk alert which inform about allegedly toxic, metabolically unstable, chemically reactive fragments present in the structure, and leadlikeness and synthetic accessibility have been investigated. In addition, the bioavailability radar was performed by visualizing the pink area that represents the optimal range of each property as plotted: lipophilicity (LIPO): −0.7 < XLOGP 3 < +5.0; SIZE: 150 g/mol < MW < 500 g/mol; POLAR (polarity): 20Å2 < topological surface area (TPSA) < 130Å2; INSOLU (insolubility): 0 < log S (ESOL) < 6; INSATU (insaturation): 0.25 < fraction of Csp3 < 1; and FLEX (flexibility): 0 < number of rotatable bonds < 9. Such molecule may be achieved as druglikeness if the pink colored zone of the radar plot of the molecule has to fall entirely in the red zone.
Antimicrobial activities
All the examined compounds were assayed for their antimicrobial activities against three Gram-positive [Bacillus subtilis JN 934392, Bacillus cereus JN 934390, and Staphylococcus aureus American Type Culture Collection (ATCC) 6538] and two Gram-negative (Salmonella entericsero type Enteritidis ATCC43972 and Escherichia coli ATCC 25922) bacterial strains and two fungal strains (Fusarium oxysporum and Fusarium phyllophilum AB 587006). The inhibition zone diameter (IZD), minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC), and minimum fungicidal concentration (MFC) were assessed based on the same protocol as described by Ghannay et al. (2020a). About 40 ml of the used agar was poured into Petri dishes. After solidification, 150 ml of the bacterial suspension was speared on the surface. After 5 minutes of contact, the wells with a diameter of 6 mm were excavated in the agar, and each well was filled with 80 μl of each extract (75 mg/ml). Finally, the Petri dishes were incubated in an oven at 37°C for 48 hours.
α-Glucosidase inhibition assay
All targets were tested for their α-glucosidase inhibitory activity using the same protocol as done by Ghabi et al. (2020). The % inhibition was determined as follows: % inhibition = [(Abscontrol−Abssample)/Abscontrol] × 100, and the IC50 values were obtained from the nonlinear regression curve.
Statistical analysis
All assays were carried out in triplicates, and the results were reported as mean ± standard error calculated by Microsoft Excel 2013.
RESULTS AND DISCUSSION
Antimicrobial properties
As can be seen from the summarized outcomes (Table 1), the α-amino-γ-lactones 1–4 derivatives showed a good-to-moderate antibacterial potencies with the most potent activity observed for compound 2 against S. aureus (IZD = 19.00 ± 0.00 mm, MIC = 3.17 mg/ml, and MBC = 6.25 mg/ml) and B. subtilis (IZD = 23.00 ± 0.88 mm, MIC = 6.25 mg/ml, and MBC = 12.50 mg/ml) compared to standard drug chloramphenicol. The inhibitory effect of isoxazolidine derivatives 5–9 was similar or even better than standard drugs for compounds 9, 5, and 6 against S. aureus, for 9 and 5 against B. subtilis, and for 9 against B. cereus. Toward Gram-negative strains, these compounds displayed moderate activities. The oxazine derivative 10 has shown a higher potency antimicrobial effect against S. aureus and B. cereus, very close activity toward B. subtilis, E. coli, and S. enteritidis strains. Regarding the γ-hydroxy-α-amino acid compounds 11–14, they exhibited better antibacterial properties than the standard drug chloramphenicol with compound 12 which was identified as the most potent, especially against S. aureus followed by those 13, 11, and 14 which are, respectively, 1.58, 1.47, 1.17, and 0.94 folds higher than chloramphenicol. In addition, compounds 12 and 13 were proved to be more susceptible than 11 and 14 against E. coli and S. enteritidis. Compound 15 exhibited a moderate-to-mild activity against the tested strains. According to the obtained IZD, MIC, and MFC values (Tables 1 and 2), it was found that all the tested compounds exerted varying degrees of inhibition against F. oxysporum and F. phyllophilum (except compound 4 for F. phyllophilum), in which some of them are good and some are moderately active when compared with the positive control, cycloheximide, with the best inhibitory activity was ascribed for compounds 12 and 13. The detail results are shown in Table 1.
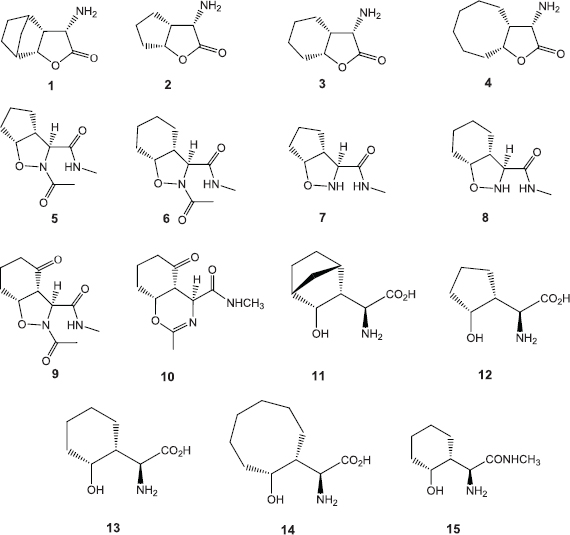 | Scheme 1. Structure of compounds 1–15. [Click here to view] |
The results of this study showed that the majority of the compounds displayed a higher bactericidal effect, especially with Gram-positive bacteria in comparison to those Gram negative. Some of them exerted a fungicidal effect against F. oxysporum (compounds 2, 5–7, and 9–14) and F. phyllophilum (compounds 2, 5–6, and 9–13). The rest are bacteriostatic or fungistatic.
In vitro α-glucosidase inhibitory activity
The results presented in Table 1 showed that the IC50 values of all compounds are in the range of 30.1 ± 0.6–256.2 ± 0.5 μM when compared with the positive control acarbose (IC50 = 780.4 ± 0.3 μM). Among them, compounds 10, 9, 5, and 6 exhibited the strongest inhibitory activities which are, respectively, about 26, 22, 17, and 14 times higher than the reference, acarbose.
Structure–activity relationships (SARs) of the synthesized compounds
The structure–activity relationship study of the tested compounds has shown that the antimicrobial potency of the title compounds undergoes the following order: α-γ-hydroxy-α-amino acids 11–14 followed by isoxazine 10, isoxazolidines 5–9, and in less degree α-amino-γ-lactones 1–4. It is worth mentioning that the variation in the susceptibility of the examined compounds against the different tested strains depends either on the cell wall of microorganisms or the structure of used derivatives which explain the effectiveness degree of each one. In fact, the cellular wall of Gram strains is formed by a thick lipoprotein–lipopolysaccharide layer which covered a thin layer of peptidoglycan and is very susceptible to the examined cycloalkylglycine derivatives. For α-amino-γ-lactone derivatives 1–4, the antimicrobial activity enhanced as the size of saturated carbocyclic ring attached to the lactone moiety decreased with the lowest one, which was observed with (1S,4S)-bicyclo[2.2.1]heptyl and cycloheptyl group, suggesting that a longer carbon chain does not promote activity. On the other hand, regarding the isoxazolidines series 5–9, the powerful antimicrobial activity was found for all compounds in comparison with that of α-amino-γ-lactone series, owing to the existence of heterocyclic nucleus. Comparing compounds 5 and 6 with those 7 and 8, respectively, it was found that the presence of acetyl substituent attached to the nitrogen (5 and 6) seems to intensify the antimicrobial profile which is in well agreement with the finding of Chawla et al. (2010). Furthermore, when we compare compound 9 with oxo-group attached to the saturated carbocyclic ring, to compound 8, we show an increase in the inhibition effect due to its withdrawing exerted effect. In addition, the presence of toxophoric –N=C–O– linkage group (compound 10) enhanced the antimicrobial activity in comparison with compound 9, especially against bacterial strains, due to their facility to react with the nucleophilic centers of the microbial cells (Zheng et al., 2018). Comparing γ-hydroxy-α-amino acid derivatives to those of α-amino-γ-lactones, we can conclude that the ones with carboxylic acid group as well as the hydroxycarbocyclic ring exhibited a better inhibitory activity against the examined strains. The highest antimicrobial potency of compound 12 may be explained by the lowest size of saturated carbocyclic ring attached to the α-amino acid moiety as compared to those 13, 14, and 11.
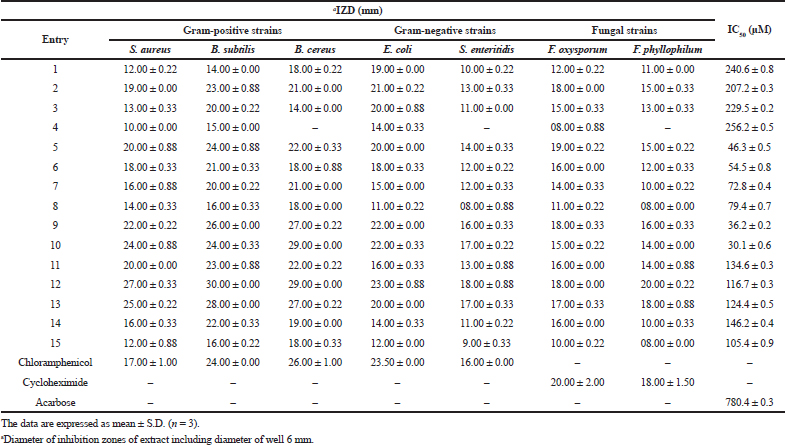 | Table 1. Antimicrobial (IZD) and α-glucosidase (IC50) inhibitory activity of the synthesized compounds. [Click here to view] |
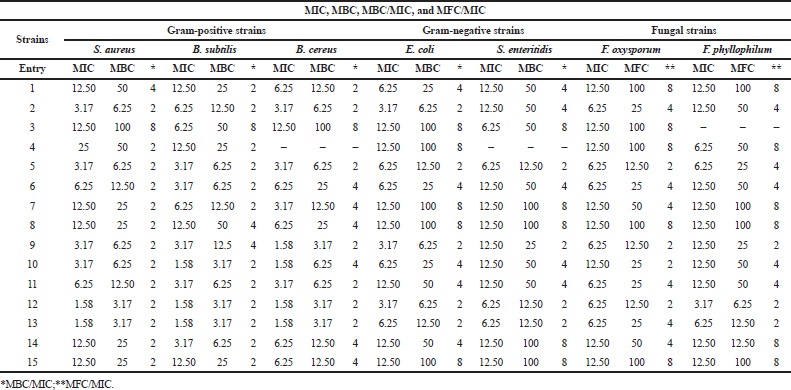 | Table 2. Determination of MIC, MBC, MBC/MIC, and MFC/MIC of the synthesized compounds. [Click here to view] |
To optimize the α-glucosidase inhibitory potential of the designed scaffold, we are interested to study their SARs. Indeed, the best inhibitory effect was observed with isoxazine (IC50 = 30.1 μM), followed by isoxazolidines (36.2 μM < IC50 < 79.4 μM), α-γ-hydroxy-α-amino acids (105.4 μM < IC50 < 146.2 μM), and α-amino-γ-lactones (207.2 μM < IC50 < 256.2 μM). The higher inhibitory activity of compound 10 can be attributed to the presence of oxazine group. In isoxazolidines series, compound 9 with both acetyl and oxo-as electron withdrawing groups attached, respectively, to the nitrogen and saturated carbocyclic ring have displayed the strongest inhibitory activity against α-glucosidase enzyme, followed by 5, in which the saturated carbocyclic ring did not have attached oxo-group. As the size of the carbocyclic ring increased, the activity was reduced (5 vs. 6 and 7 vs. 8). The removal of acetyl group (compounds 7 and 8) in comparison with those 5 and 6 also decreased the inhibitory activity, respectively. The inhibitory activity of α-amino-γ-lactones derivatives is due essentially to the presence of aminolactone moiety with the most potent inhibitory effect ascribed to compound 2 which increased generally with decreasing the size of the present carbocyclic ring.
Physicochemical, bioactivity score, and pharmacokinetic profiles
The synthesized compounds were subjected to the prediction of their ADME properties such as oral bioavailability, pharmacokinetic parameters, and the toxicity risks using an in silico Molinspiration and SwissADME tools. We explore the concept of druglikeness to reduce the failure in clinical trials.
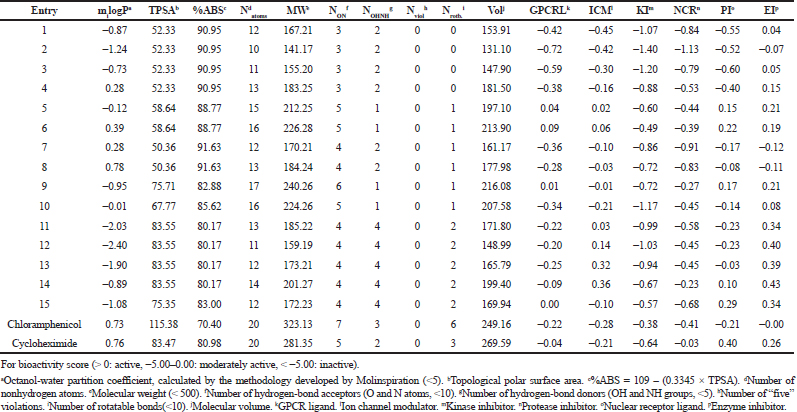 | Table 3. ADME properties of compounds 1–15 according to pre-ADMET software. [Click here to view] |
Physicochemical properties
As can be seen from the results shown in Table 3, there are no violations of Lipinski’s rule of five, and therefore, all the tested molecules are expected to be viable drug candidates. The low molecular weights revealed that all molecules are likely to be readily diffused, absorbed, or transported across cell membrane compared to bulky drug molecules. LIPO (miLogP) values of the title molecules range between −2.40 and 0.78, outlining various possible biological sites with higher hydrophilicity. The TPSA values of the selected compounds were ≤83.55 Å, suggesting a good intestinal absorption and BBB penetration, which also make compounds 6 and 7 (with the lowest TPSA level 50.36 Å) as a promising drug candidate for further research and development. The %ABS of the title compounds demonstrate high %ABS (≥80%), which is an indicator for good oral absorption. Usually, the compounds having more than 80% absorption should possess a good passive oral absorption and can be considered as a good drug. The number of rotatable bonds of the synthesized compounds is in the range 0 ≤ Nrotb ≤ 2, pointing out their very low conformational flexibility which makes them potentially bioavailable by an oral route. A number of hydrogen bond donors NOHNH (1 ≤ NOHNH ≤ 4) and hydrogen bond acceptors NON (3 ≤ NON ≤ 6) are in good accordance with the Lipinski’s rule of five and, therefore, reinforce their oral druglikeness properties.
Bioactivity score
The results are analyzed as shown in Table 3, and it was observed that all the evaluated compounds exhibited poor interaction with Kinase inhibitor (KI), poor-to-moderate interaction with nuclear receptor ligand, poor-to-higher interaction with G protein-coupled receptors (GPCRL) and Protease inhibitor (PI), and moderate-to-higher interaction with ICM and Enzyme inhibitor (EI). Interestingly, we note that the most promising compounds as per the bioactivity scores were identified to be 6 > 5 > 9 which are predicted to act by more than three mechanisms. The identified compounds were in applicable domain of bioactivity score as chloramphenicol and cyclohexamide for all drug targets.
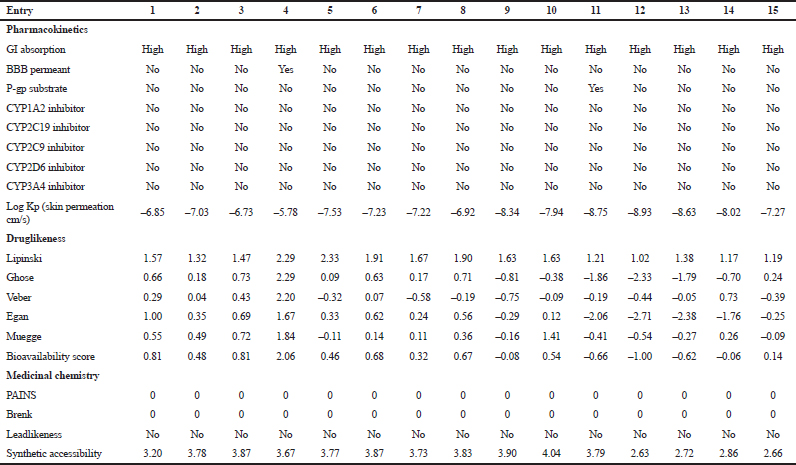 | Table 4. Pharmacokinetics, medicinal chemistry and druglikeness of compounds 1–15 according to SwissADME software. [Click here to view] |
Pharmacokinetic druglikeness and medicinal chemistry properties
The pharmacokinetic characteristics (Table 4) state that all the targets are predicted to possess good GI absorption, are not P-gp substrates, and therefore, do not cause any problem in the excretion of drug and also not able to inhibit CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, suggesting that they have no toxic ADME properties without accumulation of drug/metabolites. Their negative log Kp values are in the range from −8.93 (12) to −5.78 (4) indicating their lowest ability to penetrate through the skin. Druglikeness parameters of the title compounds can be easily assessed from bioavailability radars as expressed in pink area by the optimal range for each property (LIPO, size, polarity, solubility, saturation, and flexibility). The results shown in Table 4 indicated that all analogs displayed an excellent oral bioavailability with a bioavailability score of 0.55. Testing five druglikeness methods, all the designed drug molecules follow completely the Lipinski’s (for Pfizer), Egan’s (for Pharmacia), and Veber’s (for GlaxoSmithKline) rules; however, only compounds 1, 4–6, 8, 10–11, and 13–15 as well as 6, 9–10, and 14 do not violate Ghose’s (for Amgen) and Muegge’s (for Bayer) rules, respectively. The medicinal chemistry properties of these compounds have been predicted, and the results (Table 4) revealed that the synthetic accessibility scored approximately from 2.5 to 4.0, indicating that all the compounds possess good feasibility to be synthesized. All compounds return any pan-assay interference compounds (PAINS) and Brenk alerts. The polygon of physicochemical space (Fig. 1) showed that all parameters fall in the optimal range, and therefore, all the compounds possess a good oral bioavailability better than the tested standards.
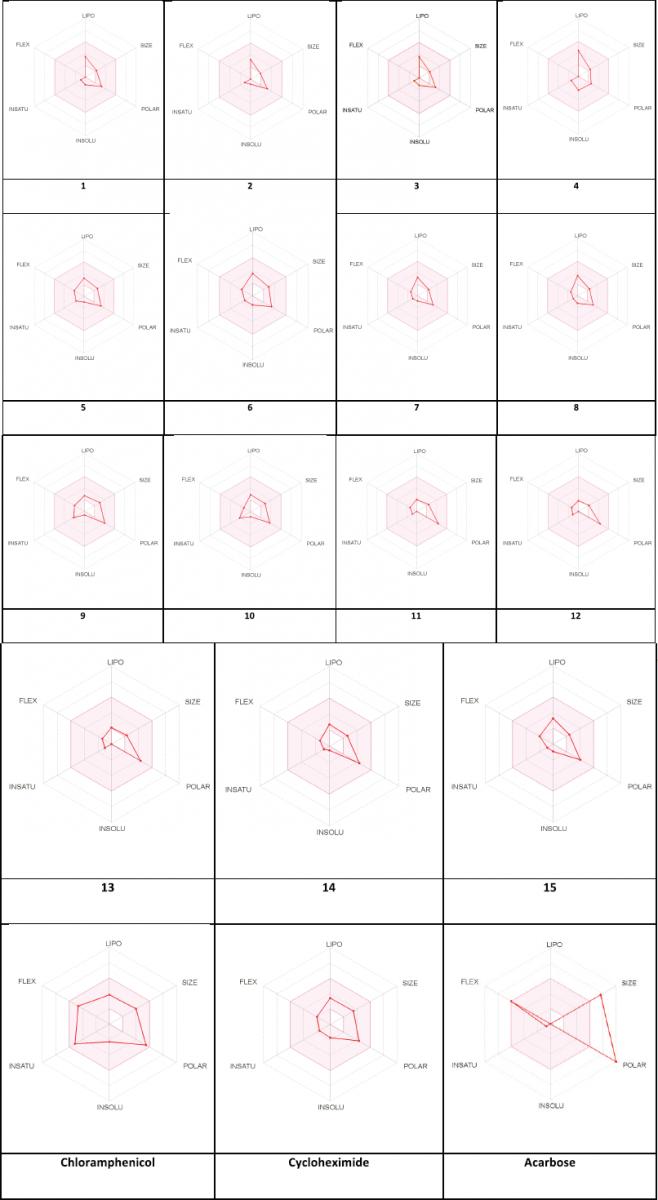 | Figure 1. Bioavailability radar of compounds (1–15) using Swiss ADME predictor. The pink area represents the optimal range for each property (LIPO, size, polarity, solubility, saturation and flexibility). [Click here to view] |
CONCLUSION
In summary, a series of enantiopure cycloalkylglycines derivatives have been evaluated for their antimicrobial and α-glucosidase inhibitory activity. All targets showed a promising antimicrobial activity against different pathogenic strains with the most potent activity was observed, essentially toward Gram-positive strains. Among the different series, γ-hydroxy-α-amino acids exhibited the strongest inhibitory effect followed by oxazine and isoxazolidines compounds and in less degree α-amino-γ-lactones, respectively. Regarding the α-glucosidase inhibitory activity, the results show that the synthesized compounds displayed a significant activity with most potent activity, which was allowed to isoxazine followed by isoxazolidines, γ-hydroxy-α-amino acids, and α-amino-γ-lactones. The physicochemical and ADME parameters show that the reported analogs have a good oral bioavailability and, therefore, could be favorable hit candidates after a systematic in vivo analysis for the further drug discovery of new antimicrobial and antidiabetic agents.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
FINANCIAL SUPPORT
None.
REFERENCES
Abda H, Aouadi K, Perrin L, Msaddek M, Praly JP, Vidal S. Stereoselective synthesis of enantiopure cycloalkylglycines by 1,3-Dipolar cycloaddition of a chiral nitrone to cycloalkenes. Eur J Org Chem, 2014; 27:6017–24. CrossRef
Abda H, Aoudi K, Msaddek M, Vidal S. Synthesis of some isoxazolidine and isoxazoline derivatives using nitrone-derived (-)-menthone via 1,3-dipolar cycloaddition with alkenes, alkynes and cycloalkenes. Heterocycles, 2016; 92:1963–75. CrossRef
Aouadi K, Jeanneau E, Msaddek M, Praly JP. 1,3-Dipolar cycloaddition of a chiral nitrone to (E)-1,4-dichloro-2-butene: a new efficient synthesis of (2S,3S,4R)-4- hydroxyisoleucine. Tetrahedron Lett, 2012a; 53:2817–21. CrossRef
Aouadi K, Jeanneau E, Msaddek M, Praly JP. Analogues of insulin secretagogue (2S,3R,4S)-4-hydroxyisoleucine: synthesis by 1,3-dipolar cycloaddition reactions of chiral nitrones to alkenes. Tetrahedron Asymm, 2008; 19:1145–52. CrossRef
Aouadi K, Jeanneau E, Msaddek M, Praly JP. New synthetic routes toward Enantiopure (2S,3R,4R)-4-Hydroxyisoleucine by 1,3-Dipolar Cycloaddition of a chiral nitrone to C4 Alkenes. Synthesis, 2007; 21:3399–405. CrossRef
Aouadi K, Lajoix AD, Gross R, Praly JP. Multi-step synthesis and biological evaluation of analogues of insulin secretagogue (2S,3R,4S)-4-Hydroxyisoleucine. Eur J Org Chem, 2009; 1:61–71. CrossRef
Aouadi K, Msaddek M, Praly JP. Cycloaddition of a chiral nitrone to allylic motifs: an access to enantiopuresugar-based amino acids displaying a stable glycosidic bond and to 4(S)- 4-hydroxy-L-ornithine. Tetrahedron, 2012b; 68:1762–8. CrossRef
Aouadi K, Vidal S, Msaddek M, Praly JP. Cycloadditions of chiral nitrones to racemic 3-substituted butenes: a direct access with kinetic resolution to enantiopure dihydroxylated amino acids. Synlett, 2006; 19:3299–303. CrossRef
Aouadi K, Vidal S, Msaddek M, Praly JP. Stereoselective synthesis of 1,2,3-triazolyl-functionalized isoxazolidines, via two consecutive 1,3-dipolar cycloadditions, as precursors of unnatural amino acids. Tetrahedon Lett, 2013; 54:1967–71. CrossRef
Brahmi J, Aouadi K, Msaddek M, Praly JP, Vidal S. A stereoselective method for the synthesis of enantiopure 3-substituted 4-hydroxyproline derivatives via 1,3-dipolar cycloadditions. CR Chimie, 2016a; 19(8):933–5. CrossRef
Brahmi J, Ghannay S, Bakari S, Kadri A, Aouadi K, Msaddek M, Vidal S, Unprecedented stereoselective synthesis of 3-methylisoxazolidine-5-aryl-1,2,4-oxadiazoles via 1,3-dipolar cycloaddition and study of their in vitro antioxidant activity. Synth Commun, 2016b; 46:2037–44. CrossRef
Cecioni S, Aouadi K, Guiard J, Parrot S, Strazielle N, Blondel S, Ghersi-Egea JF, Chapelle C, Denoroy L, Praly JP. Novel routes to either racemic or enantiopure α-amino-(4-hydroxy-pyrrolidin-3-yl)acetic acid derivatives and biological evaluation of a new promising pharmacological scaffold. Eur J Med Chem, 2015; 98:237–49. CrossRef
Chawla R, Arora A, Parameswaran MK, Sharma PC, Michael S, Ravi TK. Synthesis of novel 1,3,4-oxadiazole derivatives as potential antimicrobal agents. Acta Pol Pharm, 2010; 67:247–53.
Chiacchio MA, Giofrè SV, Romeo R, Romeo G, Chiacchio U. Isoxazolidines as biologically active compounds. Curr Org Synth, 2016; 13:726–49. CrossRef
Chougale AD, Ghadyale VA, Panaskar SN, Arvindekar AU. Alpha glucosidase inhibition by stem extract of Tinospora cordifolia. J Enzyme Inhib Med Chem, 2009; 24:998–1001. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:42717. CrossRef
Ghabi A, Brahmi J, Alminderej F, Mesaaoudi S, Vidal S, Kadri A, Aouadi K. Multifunctional isoxazolidine derivatives as α-amylase and α-glucosidase inhibitors. Bioorg Chem, 2020; 98:103713. CrossRef
Ghannay S, Alminderej F, Msaddek M, Praly JP, Aouadi K. Unprecedented Synthesis of 4-Ylidene (3S)-3-hydroxy-1-aryl (alkyl)-pyrrolidine-2,5-diones Based on 1,3-Dipolar Cycloaddition of a Sugar-derived Nitrone to N-Substituted Maleimides. Chem Select, 2019; 4:5628–32. CrossRef
Ghannay S, Bakari S, Ghabi A, Kadri A, Msaddek M, Aouadi K. Stereoselective synthesis of enantiopure N-substituted pyrrolidin-2,5-dione derivatives by 1,3-dipolar cycloaddition and assessment of their in vitro antioxidant and antibacterial activities. Bioorg Med Chem Lett, 2017; 27:2302–7. CrossRef
Ghannay S, Bakari S, Msaddek M, Vidal S, Kadri A, Aouadi K. Design, synthesis, molecular properties and in vitro antioxidant and antibacterial potential of novel enantiopure isoxazolidine derivatives. Arab J Chem, 2020a; 13:2121–31. CrossRef
Ghannay S, Kadri A, Aouadi K, Synthesis, in vitro antimicrobial assessment, and computational investigation of pharmacokinetic and bioactivity properties of novel trifluoromethylated compounds using in silico ADME and toxicity prediction tools. Monatsh Chem, 2020b; 151:267–80. CrossRef
Pili R, Chang J, Partis RA, Mueller RA, Chrest FJ, Passaniti A. The alpha-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res, 1995; 55:2920–6.
Rawlings AJ, Lomas H, Pilling AW, Lee MJ, Alonzi DS, Rountree JS, Jenkinson SF, Fleet GW, Dwek RA, Jones JH, Butters TD. Synthesis and biological characterisation of novel N-alkyl-deoxynojirimycin alpha-glucosidase inhibitors. ChemBioChem, 2009; 10:1101–5. CrossRef
Vichayanrat A, Ploybutr S, Tunlakit M, Watanakejorn P. Efficacy and safety of volibose in comparison with acarbose in type 2 diabetic patients Diabetes Res Clin Pract, 2020; 55:99–103.Zheng Z, Liu Q, Kim W, Tharmalingam N, Fuchs BB, Mylonakis E. Antimicrobial activity of 1,3,4-oxadiazole derivatives against planktonic cells and biofilm of Staphylococcus aureus. Future Med Chem, 2018; 10:283–96. CrossRef
Zhou H, Meng L, Yin X, Liu Y, Xu G, Wu J, Wu M, Yang L. Artificial biocatalytic cascade with three enzymes in one pot for asymmetric synthesis of chiral unnatural amino acids. Eur J Org Chem, 2019; 38:6470–7. CrossRef
Zitzmann N, Mehta AS, Carrouée S, Butters TD, Platt FM, McCauley J, Blumberg BS, Dwek RA, Block TM. Imino sugars inhibit the formation and secretion of bovine viral diarrhea virus, a pestivirus model of hepatitis C virus: implications for the development of broad spectrum anti-hepatitis virus agents. Proc Natl Acad Sci USA, 1999; 96:11878–82. CrossRef