
INTRODUCTION
The U.S. Food & Drug Administration (USFDA) is involved in the protection of public health by ensuring that along with food, drugs, vaccines, and medical devices are safe and protective to the public when used. USFDA’s center for devices and radiological health provides device advice. This advice includes various aspects, such as laws, regulations, and policies throughout the life cycle of a product (US Food and Drug Administration, 2018a; 2018b).
Whenever USFDA finds that a manufacturer violates any regulations significantly, it intimates the manufacturer by issuing a letter. This letter of notification is called a warning letter. This USFDA warning letter states the said violation and also outlines a corrective action needed within a given timeframe (Church and Mahoney, 2018). USFDA seeks a reply from the manufacturer in the written form. It further checks to ensure that the action taken is adequate (Shetty and Saiyed, 2015). These warning letters issued usually form the basis of future interactions among the manufacturer and regulatory agencies.
For a device manufacturer, the notification of regulatory violation through a warning letter may mean a big risk to its credibility and trust among prescribers and users. A warning letter may also notify an action indicating export certifications, or approval of new drug applications. Furthermore, it creates pressure on the manufacturer in terms of loss of investor and user trust leading to a loss in revenue (US Food and Drug Administration, 2017).
A careful look in to these warning letters issued for medical devices Current Good Manufacturing Practice (cGMP) violations often provides insight into how cGMP is implemented and deficiencies therein (Teow and Siegel, 2013). For the sake of controlling the medical devices, USFDA has classified them into three classes. Class I (Non-risk devices, e.g., gloves and bandages), Class II (moderately risky devices, e.g., infusion pumps and stents), Class III (risk devices, e.g., defibrillators and pacemakers) (Wang and Giuffrida, 2016).
Once the medical device has been authorized, it enters the market. If and when an adverse event happens, the USFDA must receive a medical device report. An adverse event is either a device-related significant injury or a device—fatality anyone, including those of the general public, may submit a medical device report, but the medical device manufacturer is made mandatory by the USFDA to report the adverse event. These compulsory manufacturer reports account for 97% of the total reports submitted. The USFDA then structures the report and agrees on the next course of action: to recall the device, reclassify it, order the manufacturers, or other to redesign. Roughly, 80,000 to 120,000 adverse event reports related to devices are submitted each year. Among these, approximately 5,000 reports are obtained by Med Watch (O’Reilly et al., 2013).
Over the last decade, the medical device industry has been growing rapidly thereby introducing software programs to push down the USFDA regulations and guidelines. However, the USFDA does not have well-defined policies or regulations to deal with the new technology. Over the past few years, USFDA released new guidance on medical device technologies and a lot of changes have been made to the existing medical device guidance documents (Gogtay et al., 2011).
There are numerous warning letter literature reviews issued to clinical research, institutional review board (Bramstedt and Kassimatis, 2004; Symonds et al., 2014), and promotional claim violation (Chatterjee et al., 2012; Kamal et al., 2009: Salas et al., 2008; Symonds et al., 2014).
The USFDA has collaborated with Medical Device Consortium to implement the Case for Quality initiative. The primary objective of the Case for Quality initiative is to raise awareness of quality issues, produce high-quality medical devices and satisfy business objectives. The program is still in the developing stage and is more concerned with Quality and risk/benefit metrics (Ananth et al., 2018; US Food and Drug Administration, 2015).
Accuracy and integrity of establishment registration and drug listing data are essential for the USFDA to reduce the subjection of the public to unsafe drugs (Khoja, 2016).
Authors of this paper could locate relatively few articles on the warning letter issued for cGMP violations of medical devices. By summarizing the number of letters and types, authors have attempted to evaluate the trends in the warning letter issued to medical devices about cGMP violations between 2008 and 2018. The objective of this review is also to study major violations pertaining to different sections of medical devices.
RESEARCH METHODOLOGY
A cross-sectional study was conducted using secondary data. A total of 669 Warning letters were included in the study distribution of the same is provided in Table 1. The study was carried out for over six months. Publically available USFDA letters under the law of the freedom of Information Act sent to the pharmaceutical company were accessed from the USFDA website.
A standard data collection tool (Excel Spreadsheet) with all letters of warning issued from January 3rd, 2008 to July 25th, 2018, was developed. Letters were manually screened and the warning letters related to medical device breaches of cGMP were screened based on the letter’s subject and content.
Medical devices were the words used in primary screening. The primary screening excluded food, tobacco product, animal product and cosmetics, biological products, finished pharmaceuticals, and active pharmaceutical ingredients. The secondary screening was carried out using keywords, such as GMP (Good Manufacturing Practice), cGMP (Current Good Manufacturing Practice), Quarterly Statistical Report, and MDR (Medical Device Reporting) based on primary screening.
This work did not cover the warning letters relating to Good Clinical Practice, Good Laboratory Practice, and infringements of promotional claims.
Data was analyzed, and results were presented descriptively.
 | Figure 1. Graphical representation of warning letters related to cGMP violation issued by USFDA by years to the medical device. [Click here to view] |
 | Table 1. Sample size distribution from 2008 to 2018. [Click here to view] |
RESULTS AND DISCUSSION
Overall, 669 warning letters issued for medical device cGMP violations were reviewed between January 3, 2008, and November 9, 2018. In 2008, the FDA issued 79 letters. Between 2009 and 2013, there was a downward trend in letter number that fell to 47 in 2009, 49 in 2010, 73 in 2011, 66 in 2012 and 46 in 2013. The number of warning letters issued in 2014 was 101, followed by 106 in 2015, as the FDA focused more on data integrity issues, while the number decreased to 53, 27 and 19, respectively, in 2016, 2017, and 2018, as provided in Figure 1.
The highest number of warning letters was issued to manufacturers located in the USA (379) in the past ten years, followed by Canada (52), and China (37). Germany (27) and UK (19) were issued higher number of warning letters followed by Japan (15), France (11), and India (10) as shown in Figure 2.
From Figure 3, it can be seen that the most violated section is 820.30 which falls under subpart C of Title 21 CFR with 603 infringements. This section represents the design control requirements for Class 1, Class 2, and Class 3 products, which includes General, Design and development planning, Design input, Design output, Design review, Design verification, Design validation, Design transfer, Design changes, and Design history file.
Section 820.100 under subpart J was found to be the second most violated with 535 violations. This rule ensures that each manufacturer establishes and maintains procedures for carrying out corrective and preventive actions.
Section 820.198 under subpart M has been observed 490 times which means that the complaint files are to be established on time and maintained by each manufacturer. Responsibility of the manufacturer is to maintain the records of the details related to the investigation, corrective actions and replies given for complaint.
 | Figure 2. Graphical representation of warning letters related to cGMP violation issued by USFDA to medical device across the world. [Click here to view] |
 | Figure 3. Graphical representation of 21 CFR 820 violations for medical devices. [Click here to view] |
Section 820.50 under subpart E violations were observed for 263 times which represents Purchasing Control and ensures that each manufacturer shall establish and maintain procedures of all purchased or otherwise received products and services conforming to specified requirements. This rule makes sure that the manufacturer must evaluate the suppliers, contractors and need to be documented along with purchase data.
Section 820.80 under subpart H with 261 violations represents each manufacturer shall establish and maintain procedures for acceptance activities which includes inspections, tests, or other verification activities.
Section 820.70 and 820.75 violations were observed for 260 and 256 each. These sections fall under subpart G in which section 820.70 deals with Production and process control including SOP’s, process parameters, compliance with specified reference standards, approval processes, and process equipment. In case 820.75 deals with process validation which ensures that validation must be done with some qualified personnel. In case of any deviation, the entire process must have revalidated and documented (Table 2).
From Figure 3, it is observed that Section 803.17 of Title 21 CFR is most violated with 178 violations which represent the requirements for developing, maintaining, and implementing written MDR procedures.
From the Figure 4, it can be observed that Class 2 type of medical devices which are of moderate risk were found to be most violated (82%) followed by Class 3 devices having the Highest risk with 7% which requires Pre-Market Approval (PMA). The PMA process is substantially longer than the 510(k) process and requires extensive clinical trials to demonstrate the safety, efficacy, and intended use of the device.
From Figure 5, we can observe that only 32% of the companies responded to the warning letters in a speculated period which was issued by USFDA although 52% Not Issued the closeout letter which means the companies did not respond to the USFDA’s warning letter. 16% of the letters were considered as non-applicable letters, i.e., warning letters issued by the USFDA before 2008.
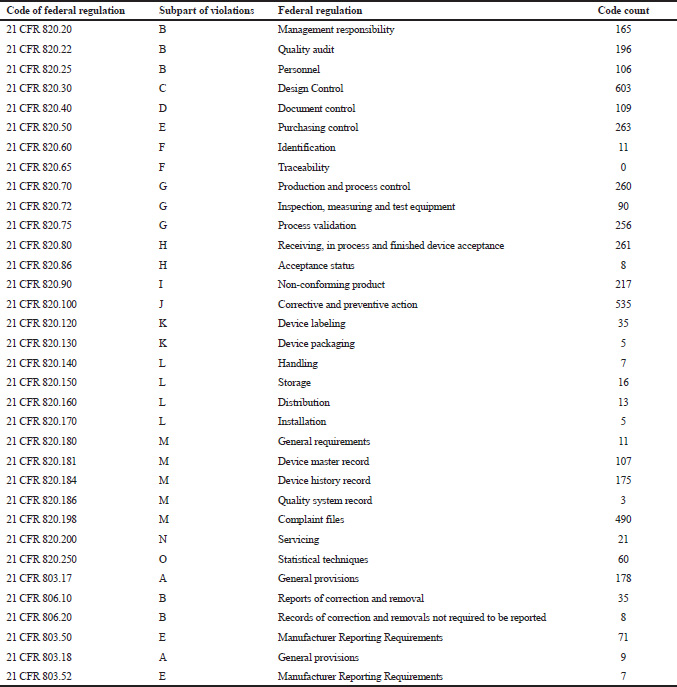 | Table 2. Count of warning letters issued to the medical device for 21 CFR 820 violations. [Click here to view] |
 | Figure 4. Graphical representation of the percentage of warning letters issued to a different class of medical device. NA: Not Applicable, NI: Not Issued. [Click here to view] |
 | Figure 5. Graphical representation of the percentage of closeout letter issued by the companies. [Click here to view] |
CONCLUSION
With the time, scientific developments, and increased awareness of both regulatory authorities and industries/academic organizations, overall improvement is observed with the significant number of decreased warning letters with the time. This improvement is reflected in the lower incidence rates on patient safety/toxicities and improvement in the quality of medicines. Ultimately, all of the variables mentioned above are likely to have attributed to the recent decline in Warning letters. There are likely to be many more modifications in device enforcement as USFDA reinforces its role in recent device technologies.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
ETHICAL APPROVAL
Not required.
REFERENCES
Ananth L, Gurbani NK, Kumar S, Gujavarti B. A retrospective study of Warning Letters issued by US FDA over 2015–2017. Int J Drug Regul Aff, 2018; 6(2):48–53. CrossRef
Bramstedt KA, Kassimatis K. A study of warning letters issued to institutional review boards by the United States Food and Drug Administration. Clin Invest Med, 2004; 27(6):316–23.
Chatterjee S, Patel HK, Sansgiry SS. An analysis of the warning letters issued by the FDA to pharmaceutical manufacturers regarding misleading health outcomes claims. Pharm Pract, 2012; 10(4):194–8. CrossRef
Church R, Mahoney S. Trend in FDA good manufacturing practice warning letters. RAJ Pharma, 2009; (20):433–5.
Gogtay NJ, Doshi BM, Kannan S, Thatte U. A study of warning letters issued to clinical investigators and institutional review boards by the United States Food and Drug Administration. Indian J Med Ethics, 2011; 8(4):211–4. CrossRef
Kamal KM, Desselle SP, Rane P, Parekh R, Zacker C. Content analysis of FDA warning letters to manufacturers of pharmaceuticals and therapeutic biologicals for promotional violations. Drug Inf J, 2009; 43(4):385–93. CrossRef
Khoja FS. A review on USFDA warning letter and violation observed in Pharmaceutical Industry. PharmaTutor, 2016; 4(12):33–6.
O’Reilly EK, Holbein MB, Berglund JP, Parrish AB, Roth MT, Burnett BK. Warning Letters to Sponsor-Investigators at Academic Health Centres–The Regulatory “Canaries in a Coal Mine”. Clinical and investigative medicine. Medecine clinique et experimentale, 2013; 36(6):E290. CrossRef
Salas M, Martin M, Pisu M, McCall E, Zuluaga A, Glasser SP. Analysis of US food and drug administration warning letters. Pharm Med, 2008; 22(2):119–25. CrossRef
Shetty YC, Saiyed AA. Analysis of warning letters issued by the US Food and Drug Administration to clinical investigators, institutional review boards and sponsors: a retrospective study. J Med Ethics, 2015; (41):398–403. CrossRef
Symonds T, Hackford C, Abraham L. A review of FDA warning letters and notices of violation issued for patient-reported outcomes promotional claims between 2006 and 2012. Value Health, 2014; 17(4):433–7. CrossRef
Teow N, Siegel SJ. FDA regulation of medical devices and medical device reporting. Pharmaceut Reg Affairs, 2013; 2(110):2. CrossRef
US Food and Drug Administration. Inspections, compliance, enforcement and criminal investigations. 2015. Available via https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/enforcement-story (Accessed 26 May 2018).
US Food and Drug Administration. Regulatory procedures manual. Chapter 4: Advisory actions 4-1 – warning letters. 2017. Available via https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-manuals/regulatory-procedures-manual (Accessed 25 June 2018).
US Food and Drug Administration. Device advice: comprehensive regulatory assistance, 2018a. Available via https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance (Accessed 25 June 2018).
US Food and Drug Administration. What Does FDA do? 2018b. Available via https://www.fda.gov/about-fda/fda-basics/what-does-fda-do (Accessed 25 June 2018).
Wang J, Giuffrida A. The development of medical devices in foreign markets. Pharmaceut Reg Aff, 2016; 5(167):2.