
INTRODUCTION
Viral infections attain greater importance throughout the globe as it impacts a higher level of mortality and morbidity. Infection such as dengue caused by the serotypes of dengue virus (DENV) with class ranges from 1 to 4. DENV often turns epidemic and becomes major threat to the public health in both tropical and subtropical regions (Halstead, 2007; Mustafa et al., 2015). The enzyme RNA-dependent RNA polymerase (RdRp) has a diversified sequence and plays a vital role in viral replication through the transcription of genomic components. The average length of the core RdRp domain is less than 500 amino acids and is folded into three subdomains, namely, thumb, palm, and fingers resembling a right-handed cup (Sangita et al., 2018).
Extensive research on immunology has hypothesized host immune response toward DENV and its mechanism of pathogenesis. The outcome of this research revealed that DENV mainly stimulates humoral immune response mediated by macrophages followed by antigen presentation and clonal expansion (Gupta et al., 2012). It was also believed that the Asian climatic condition favors the breeding habitat of the Aedes Aegypti vector (Guhathakurta et al., 2015).
Currently, there are no clinically approved standard treatments available for dengue infection. Affected individuals present a wide range of fatal clinical symptoms staring from severe abdominal pain and ending up to hemorrhage. The utilization of antiviral agents may help in reducing the bioburden of viral load in emergency cases. A viral genome undergoes constant mutation results in the change of polymorphs of the binding site and lack of affinity. Due to this reason, the conventional antiviral agent fails to provide adequate cure and restoration. Hence, dire need of alternates is of paramount importance in the case of dengue viral infestations. The Indian medicinal plant research documented the pharmacological effects of some herbs against dengue infections such as Azadirachta indica indicated for treating dengue virus type-2 (Pawan and Pooja, 2017), Carica papaya extract for its platelet rejuvenation property which is under clinical trial (Kasture et al., 2016), Brassica juncea (Das et al., 2016), Plectranthus vettiveroides (Nisheeda et al., 2016), and Solanum xanthocarpum (Smita, 2015), which are traditionally used for managing dengue fever in different parts of India. In general, herbs may act by multiple mechanisms due to its unique blend of structural versatile secondary metabolites, such as alkaloids, flavonoids, terpenes, saponins, and glycosides.
Flavonoids are a large group of hydroxylated phenols existing in a wide variety of medicinal plants. It has also been extensively studied for its efficacy in cancer, neurodegenerative disorders, and cardiovascular and metabolic diseases (Panche et al., 2016). Considering its therapeutic potential, flavonoids have been utilized in pharmaceuticals, nutraceuticals, dietary supplements, and adjuvants and also in some cosmetic preparations (Duangjai et al., 2018).
A computational analysis provides crucial information about the active binding site on the surface of the target along with residual amino acids involved in hydrogen bond formation with the functional groups, which relies on the lead molecules. With this information, researcher and synthetic chemist tend to design the pharmacophore skeleton of drug candidate. An in silico analysis clearly reflects the druglikeness property and ability to cross the biological barriers even recent tools have millions of inputs regarding LD50 values and pharmacokinetic and toxic kinetic property of the new drug entity. A recent discovery of antiviral molecules from 5.4 million drug-like compounds strongly suggests the role of virtual screening in the process of new drug discovery (Sean et al., 2014).
Enzyme inhibition potential of flavonoids makes it to be the first-line candidates in the process of new drug discovery (Gorniak et al., 2019). A structural activity relationship of apigenin belonging to flavone (4′,5,7-trihydroxyflavone), hesperidin to flavanone (6-O-(alpha-L-rhamnopyranosyl)-beta-D-glucopyranosyl moiety), kaempferol to favonol (3,4′,5,7-tetrahydroxyflavone), myricetin to favonol (3-(6-rhamnosylgalactoside), and naringenin to flavanone (4′,5,7-trihydroxy flavanone 7-rhamnoglucoside) has core functional benzene ring and spatial orientation of ketone and hydroxyl groups of these molecule expected to exert better affinity toward the active site of the selected target. The rationale for selecting the above-mentioned flavonoids relies on high structural versatility of the molecular skeleton and prominent functional side chains that could attribute to the bidding on target site that may elicit the expected biological activity. Further, still, there is no proper documentary research evidence on exploring the antiviral potential of these lead molecules against DENV. Hence, this study aimed to evaluate the RdRp enzyme inhibition potential of these bioflavonoids along with safety and pharmacokinetic profiling using suitable prediction tools.
MATERIALS AND METHODS
Protein–ligand docking
Molecular docking analysis was performed using AutoDock version 4. (https://www.dockingserver.com), which predicts the interactions between lead phytomolecules and the selected protein target [dengue virus NS5 RNA-dependent RNA polymerase (RdRp)].
Protein preparation
Three-dimensional (3D) structure of dengue virus NS5 RNA-dependent RNA polymerase with Protein Data Bank (PDB)-2J7U (Fig. 1) was retrieved from the Research Collaboratory for Structural Bioinformatics. The protein structure was cleaned by removing the existing lead components, water molecules cleaved, Gasteiger charges computed with the inclusion of polar hydrogens, and merging of nonpolar and rotatable bonds was defined using AutoDock 4 (Stefano et al., 2016).
Active site prediction on the target protein
Core amino acid involved in mediating the activity of the target RdRp was predicted using Ramachandran plot, indicating a majority of the active site amino acid residues on the B-side chain of the target enzyme. The prediction was done by MolProbity server and also through a literature survey. Ramachandran plot signifies the sequential paradigm of 573 residues which are present in refinement carried out in REFMAC 5.2.0019. R = 0.200; Rfree = 0.234, and the structure was solved at 1.85 Å resolution as shown in Figure 2.
 | Figure 1. 3D structure of NS5 RNA-dependent RNA polymerase (PDB)-2J7U. [Click here to view] |
 | Figure 2. Ramachandran plot indicating active site amino acid residues in cluster form. [Click here to view] |
Ligand model preparation
The structures of the bioactive lead compounds such as apigenin, hesperidin, kaempferol, myricetin, and naringenin were outlined using ChemDraw sketch software and converted from two-dimensional (2D) to 3D structures.
Prediction of ADME and toxicity properties
Absorption, distribution, metabolism, and elimination-related toxicities of all the selected lead phytocompounds were calculated using Swiss ADMET (absorption, distribution, metabolism, excretion, and toxicity) web tool (Daina et al., 2017). Druglikeness properties of all the leads were subjected to Lipinski’s and Ghose’s rule of druglikeness (Antoine et al., 2017). (http://www.swissadme.ch/index.php).
Docking simulations
The 3D componential structure of lead molecules and protein was docked using AutoDock analytical tool version 4. Affinity (grid) maps of ×× Å grid points and 0.375 Å spacing were generated using the AutoGrid program. AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. The docking simulations were performed using the programmed algorithm inbuilt with preautomation in the software (Osterberg et al., 2002). Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was derived from two different runs that were set to terminate after a maximum of 250,000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å and quaternion and torsion steps of 5 were applied.
RESULTS AND DISCUSSION
ADMET and druglikeness analysis
The absorption and distribution pattern of flavonoids in humans purely depends on its molecular size, functional group, and other physicochemical properties (Table 1). In general, the aglycone part of the flavonoids is biologically active on administration to the biological system, flavonoid glycosides are subjected to enzymatic action by lactase phloridzin hydrolase, and β-glucosidase enzyme prevails outside the brush border membrane of the small intestine. Subsequently, the liberated aglycone can be absorbed across the small intestine (Day et al., 2000).
Kinetic profiling offers a greater advantage of predicting the desired pharmacological activity of the leads in the process of new drug discovery. Drug with good intestinal absorption may render to achieve the expected bioavailability on administration (Louis and Harvey, 2017). Penetrability and bioavailability are the key prime factors that determine the drug action on the target site (Jay et al., 2016). It was observed from ADMET analysis that all lead molecules such as apigenin, kaempferol, myricetin, and naringenin have good gastrointestinal (GI) absorption except hesperidin with low penetration. Further, none of the compounds are permeated to cross blood–brain barrier (BBB). Skin permeation scoring of lead molecule ranges from Log Kp −5.80 cm/s to −10.12 cm/s shows that these compounds have a tendency to cross biological barriers in humans. All the selected lead compounds obey Lipinski’s and Ghose’s rule of druglikeness except hesperidin reported with three violations with Lipinski and four with Ghose (Table 2).
 | Table 1. Physicochemical properties of lead compounds. [Click here to view] |
 | Table 2. Pharmacokinetic and druglikeness profile of lead compounds. [Click here to view] |
Toxicity prediction analysis
Safety index of the compounds priorities top on the objective of preclinical and clinical phases of the new drug investigation. Compounds with high safety index reciprocate more space for optimizing the dose range finding for animal and human (Andrade et al., 2016). Toxicity prediction assessment report evidences the safety profile of apigenin, hesperidin, kaempferol, myricetin, and naringenin with respect to cytotoxicity, hepatotoxicity, carcinogenicity, immunotoxicity, and mutagenicity. A predicted score for all the lead molecules falls less than 1, which signifies the compatibility nature of such compounds for better suits therapeutic (Table 3).
LD50 data obtained from ADMET analysis warrant the safety level of all five lead compounds. The predicted values of LD50 range from 159 mg/kg to 12,000 mg/kg (Table 3), and the higher the value, the wider will be the safety margin. The predicted value of apigenin is 2,500 mg/kg, and for hesperidin, it is 12,000 mg/kg. Similarly, for kaempferol, myricetin, and naringenin, the values are 3,919 mg/kg, 159 mg/kg, and 2,000 mg/kg. The range of LD50 makes the compound suitable for varying range of titrated dose, especially with good GI absorption. It is highly possible to achieve the required level of bioavailability. Safety of the drug is of utmost important while treating acute viral infections such as dengue, as the patients are already in immune-compromised state even small adverse event may end lethally. This is because most of the pharmaceutical inventors attentively spend 60% of time in deriving safety data of their trial drugs (Sanvidhan et al., 2015).
Molecular docking analysis
Docking becomes a reliable drug discovery tools for optimizing the potential lead molecules against selective target. Docking achieves nearly 80% of simulation level which made this technique more unique in the field of drug design. Historic approval of HIV-1 protease inhibitor by the Food and Drug Administration (FDA) authorities clearly emphasizes the success rate of this analytical technique (Arodola and Soliman, 2015). The docking score implicates the binding affinity between the lead and target; higher the negativity in the value showcases the level of potency of the drug (Evanthia et al., 2014). The development and advancement in the field of computational analysis increased the precision level in identifying the potential drug molecule and deriving its mechanism of action at target site. Selective alterations in the functional groups greatly minimize the nonspecific binding and impede the adverse event at the clinical level (Ramsay et al., 2018). Compound apigenin exhibited a quite tight binding on to the RdRp enzyme with a binding energy of −8.28 kcal/mol and ranks first in the compound series. The second best score was ranked by compound kaempferol with a binding energy of −7.00 Kcal/mol, followed by the myricetin, naringenin, and hesperidin compounds with the binding energy of −4.37, −4.35, and −3.20 Kcal/mol, respectively (Table 4).
 | Table 3. Toxicity prediction analysis of lead compounds. [Click here to view] |
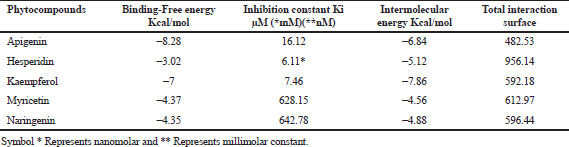 | Table 4. Summary of the molecular docking studies of the lead compounds against dengue virus NS5 RNA-dependent RNA polymerase (RdRp)—PDB 2J7U. [Click here to view] |
An inhibition constant is directly proportional to binding energy in the process of molecular docking analysis (Tatu and Antti, 2018). In this study, inhibition constant of the selected phytoflavonoids ranges from 853.19 nM to 642.78 μM. Thus, from the report, it was indicated that all the phytoconstituents have a promising RdRp enzyme inhibition activity. In virtual screening tool, intermolecular energy also considered as prime predictive factor determines the lead receptor interaction, and this study reveals the intermolecular energy of all five compounds ranging between −4.56 and −8.82 kcal/mol (Table 4). A decrease in intermolecular energy of the compounds ultimately coincides with the binding energy.
RdRP is an enzyme that catalyzes the replication of RNA from an RNA template. It catalyzes the synthesis of the RNA strand complementary to a given RNA template. Amino acids such as Arg-737, Arg-729, and Ser-710 are the important residues which have a strong influence on RNA virus initialization (Yap et al., 2007). These three residues are strictly conserved across positive-strand RNA viruses known to initiate replication using a de novo mechanism. Residues such as Ser-710 and Arg-729 (from motif E) make a hydrogen bond with the γ-phosphate. Those residues thus provide the platform for de novo RNA initiation by the viral polymerase domain.
Lead compound exhibits a strong binding on the target by forming hydrogen bonds. Mostly, hydroxyl groups of the drug candidates may involve in such interaction. It was observed from the study that therapeutic leads such as apigenin, hesperidin, and kaempferol are revealing exponentially high-level interaction out of five selected leads with core active amino acid residues (710 SER, 729 ARG, and 737 ARG) on the RdRp enzyme (Table 5 and Figs. 3–5). Lead molecule myricetin reveals partial interaction (710 SER and 729 ARG) on the active residues (Table 5 and Fig. 6). Similarly, binding behavior was exhibited by the compound naringenin on the active sites of the enzyme (729 ARG and 737 ARG) as shown in Table 5 and Figure 7.
 | Table 5. Interaction of lead compounds with active site amino acid residue of RdRp—PDB 2J7U. [Click here to view] |
 | Figure 3. Docking pose and HB plotting analysis of apigenin with dengue virus NS5 RNA-dependent RNA polymerase PDB 2J7U. [Click here to view] |
 | Figure 4. Docking pose and HB plotting analysis of hesperidin with dengue virus NS5 RNA-dependent RNA polymerase PDB 2J7U. [Click here to view] |
 | Figure 5. Docking pose and HB plotting analysis of kaempferol with dengue virus NS5 RNA-dependent RNA polymerase PDB 2J7U. [Click here to view] |
 | Figure 6. Docking pose and HB plotting analysis of myricetin with dengue virus NS5 RNA-dependent RNA polymerase PDB 2J7U. [Click here to view] |
 | Figure 7. Docking pose and HB plotting analysis of naringenin with dengue virus NS5 RNA-dependent RNA polymerase PDB 2J7U. [Click here to view] |
CONCLUSION
Still, there is no proper gold standard treatment available for curing dengue viral infection. Vaccines and other antiviral agents are currently under various clinical phases, and none of these therapeutics is of clinical beneficial for current acute infective cases. Herbal supplements either as core ingredients or in combination definitely achieve synergistic action in treating dengue. The present investigation strongly suggests that lead compounds such as apigenin, hesperidin, and kaempferol exert excellent antiviral property by inhibiting RdRp enzyme through interactive binding with 710 SER, 729 ARG, and 737 ARG active amino acids. Hence, in the future, these bioactive flavonoids may grab therapeutic importance in managing dengue infections.
ACKNOWLEDGMENT
The authors would like to acknowledge the Indian Council of Medical Research (ICMR), Government of India, New Delhi.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
Andrade EL, Bento AF, Cavalli K, Oliveira SK, Schwanke RC, Siqueira JM, Freitas CS, Marcon R. Non-clinical studies in the process of new drug development—part II: good laboratory practice, metabolism, pharmacokinetics, safety and dose translation to clinical studies. Braz J Med Biol Res, 2016; 49:1–19. CrossRef
Antoine D, Olivier M, Vincent Z. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1–13. CrossRef
Arodola O, Soliman MC. Could the FDA-approved anti-HIV PR inhibitors be promising anticancer agents? An answer from enhanced docking approach and molecular dynamics analyses. Drug Des Devel Ther, 2015; 9:6055–65. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:42717. CrossRef
Das PK, Badore NS, Patel P, Deshmukh N.Ethnomedicinal wound healing plant in Khargone district of Madhya Pradesh: a survey over Nimari communities. Pharm Biol Eval, 2016; 3:388–99.
Day AJ, Canada FJ, Diaz JC. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett, 2000; 468:166–70. CrossRef
Duangjai T, Areeya T, Apinan P, Aujana Y. Flavonoids and other phenolic compounds from medicinal plants for pharmaceutical and medical aspects: an overview. Medicines (Basel), 2018; 5:1–16. CrossRef
Evanthia L, George S, Demetrios KV, Zoe C. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem, 2014; 14:1923–38. CrossRef
Gorniak I, Bartoszewski R, Kroliczewski J. Comprehensive review of antimicrobial activities of plant flavonoids. Phytochem Rev, 2019; 18:241–72. CrossRef
Guhathakurta P, Rajeevan M, Sikka DR. Observed changes in southwest monsoon rainfall over India during 1901–2011. Int J Climatol, 2015; 35:1881–98. CrossRef
Halstead SB. Dengue. Lancet, 2007; 370:1644–52. CrossRef
Jay T, David C, Ryan G, Alison S, Leslie AK. Key factors influencing ADME properties of therapeutic proteins: a need for ADME characterization in drug discovery and development. MAbs, 2016; 8:229–45. CrossRef
Kasture PN, Nagabhushan KH, Kumar A. A multi-centric, double-blind, placebo-controlled, randomized, prospective study to evaluate the efficacy and safety of Carica papaya leaf extract, as empirical therapy for thrombocytopenia associated with dengue fever. J Assoc Phys India, 2016; 64:15–20.
Louis L, Harvey W. Predicting oral drug absorption: mini review on physiologically-based pharmacokinetic models. Pharmaceutics, 2017; 9:1–14. CrossRef
Mustafa MS, Rasotgi V, Jain S. Discovery of fifth serotype of dengue virus (DENV-5): a new public health dilemma in dengue control. Med J Armed Forces India, 2015; 71:67–70. CrossRef
Nisheeda BA, Safeer PM, Sreekumar S, Biju CK, Seeja G. A review on Plectranthus vettiveroides: an endemic to South Indian high value aromatic medicinal plant. IOSR J Pharm Biol Sci, 2016; 11:1–11.
Gupta N, Srivastava S, Jain A, Umesh C. Dengue in India. Indian J Med Res, 2012; 136:373–90.
Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins, 2002; 46:34–40. CrossRef
Panche, AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci, 2016; 5:1–15. CrossRef
Pawan KS, Pooja R. Evolving herbal formulations in management of dengue fever. J-AIM, 2017; 8:207–10. CrossRef
Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med, 2018; 7(1):3. CrossRef
Sangita V, Burra VLS, Ramasamy S. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses, 2018;10:1-23. CrossRef
Sanvidhan GS, Prabhat K, Harsh N.Role of Pharmacovigilance in India: an overview. Online J Public Health Inform, 2015; 7:223. CrossRef
Sean E, Joel SF, Megan C. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000 Res, 2014; 3:277. CrossRef
Smita S.Indigenous medicinal plants used for treatment of dengue fever by Tribals of Chhattisgarh (India). Int J Pharm Bio Sci, 2015; 6:404–10.
Stefano F, Ruth H, Michael EP, Michel S, David SG, Arthur J. Olson. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc, 2016; 11:905–19. CrossRef
Tatu P, Antti P. Binding affinity via docking: fact and fiction. Molecules, 2018; 23:1–11. CrossRef
Yap TL, Xu T, Chen YL. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol, 2007; 81:4753–65. CrossRef