
INTRODUCTION
Cancer is one of the dreadful, serious, and life-threatening diseases that cause a serious harm to human health. It is the uncontrolled and rapid growth of abnormal cells and the most serious afflictions in the world. Cancer represents one of the leading reasons for death worldwide. According to a survey, one in eight deaths worldwide is occurring due to cancer (Cancer Statistics, 2007). Cancer has become the second most leading cause of death around the world, and approximately 9.6 million deaths have been reported in the year 2018. WHO (2018) evident the cases of liver, stomach, prostate, colorectal, and lung cancers in males; moreover, the cases of thyroid, lung, cervix, colorectal, and breast cancers prevail most commonly in females. Although concerning the treatment of cancer, various discoveries have been made worldwide but a complete cure still proposes a challenge. Evident shows that even after understanding the role of various anticancer agents and the effectiveness of these drugs, there is still a significant research effort that has to be done in the development of such target drug delivery systems and new management therapies for the improvement of both treatment and diagnosis to enhance the safety and efficacy of the therapeutic drugs which makes this a center of research work everywhere (Bae and Park, 2011; Zhu et al., 2013).
Nowadays, a molecular hybrid approach of merging two potential pharmacophores in one molecule has become one of the best effective strategies to beat this deadliest disease. Hence, in the present study, we have discussed all the recently reported hybrids of Uracil or 5-Fluorouracil (5-FU) that exclusively have anticancer properties. This study focuses to assist the medicinal chemists across the world in their molecular designing, who are putting their constant efforts in the research and development of novel hybrid anticancer molecules.
Hybrid molecules
Cancer is a very complex disease and cannot be easily treated with a single drug or with combinations of drugs. Therefore, chemotherapy targets the administration of a high dose of single drug and combination of drugs that led to critical side effects and drug–drug interactions, which has to be minimized. Therefore a technique known as molecular hybridization was discovered that offered a path to overcome these barriers. This technique is based on the inculcation of two or more active compounds into one molecule without altering their original therapeutic value (Bae and Park, 2011). This technique has been proven productive in the development of various synthetic hybrids that have been found to be more medically effective than their individual components due to their improved affinity and efficacy toward the target sites. Moreover, in this era, molecular hybridization has been proven to be a boon for the designing and development of new and potential antitumor agents (Xu et al., 2019).
Types of hybrid molecules
Molecular hybridization techniques (Fig. 1) can be classified as follows:
Linking technique
This technique focuses on linking the two pharmacophores via a flexible linker or spacer. The linkers could be breakable or non-breakable based upon the conditions prevailing at the site of action, that may or may not impart its own therapeutic significance. Cleavable linkage releases the active part of drug at their specific target site whereas non-cleavable linkage retains both pharmacophores until therapeutic action has been achieved.
Fusing technique
In this, preparation of molecular hybrids is done via covalently linking two bioactive pharmacophores; usually, a condensation reaction between the functional groups of each component is involved in the synthesis of these hybrids.
Merging technique
Merge hybrids must have some common parts in their structure. They are small in size as compare to hybrids containing linker and fused hybrid. Usually these hybrids are synthesized by a cyclization reaction between either two acyclic components or between one cyclic and one acyclic component.
All the above-mentioned techniques are being utilized for the synthesis of novel potential hybrids based upon the required pharmacokinetic parameter. Hybrid drug therapy is always found to be better than a single drug therapy because of the advantages (Kucuksayan and Ozben, 2019), which are summarized as follows:
- The ideal timing window for the hybrid to act upon
- Enhanced synergistic action of the hybrids in comparison to the concurrent use of separate drugs
- Attaining limited risk of drug resistance due to its incorporation in hybrid molecule
- Improved pharmacokinetic features of the hybrids
- Diminished toxic adverse effects as compared to the administration of multiple agents
- Improved patient compatibility toward treatment
- Lowering the cost of `treatment
Designing strategy of hybrid molecules
For the determination of the safe and effective cure, whole scientific community is putting their constant efforts for the development of potential hybrid molecules that possess unique anticancer properties. The reported studies evident the continuous ongoing research in design and development of potent anticancer drugs via novel chemical and biochemical strategies. The hybrid molecules are basically designed by two major strategies (Fortin and Bérubé, 2013).
Strategy I (Merging haptophoric moieties of different drugs)
In this approach, haptophoric moieties of different drugs are merged to obtain new anticancer hybrids. Combination of haptophoric moieties is done in such a way that the molecular structure retains its affinity and potency until it reaches to its biological target.
This approach is further divided into two sub-categories as depicted in Figure 2.
Blending two drugs having the same target site
In this approach, haptophoric moieties of different drugs are merged to obtain new anticancer hybrids. Combination of haptophoric moieties is done in such a way that the molecular structure retains its affinity and potency until it reaches to its biological target.
Indenoisoquinoline-camptothecin hybrids
These hybrids are obtained by blending two Topoisomerase I inhibitors indenoisoquinoline and camptothecin(1). The hybrids showed good antiproliferative activity in the micro-molar range but they possess lower biological activity than Camptothecin and 5, 11 diketoindenoisoquinoline as parental compounds alone (Fox et al., 2003).
 | Figure 1. Different types of molecular hybrids. [Click here to view] |
 | Figure 2. Hybrid strategy by merging haptophoric moieties of drugs. [Click here to view] |
Epoxyfuroacridone
These hybrids are obtained by blending Acridone moiety present in Acronycine and Epoxyfuran moiety present in Psorospermin. This Epoxyfuroacridone (2) exhibits antiproliferative activity in the nanomolar range (Nguyen et al., 2009).
Blending two drugs having different target site
The hybrid drugs can be obtained by fusing those molecules which act through different targets or through a different mechanism. These hybrids are blend in such a flexible way that molecule can access easily to each target and counter attack over target achieved.
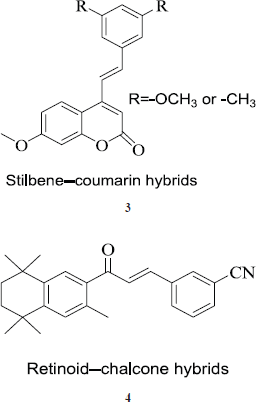
Stilbene-coumarin hybrids
These hybrids are formed by a combination of phenylethenyl moiety of resveratrol with the coumarin ring system (3); both are reported to have anticancer properties. Moreover, both the moieties act on the same target but via different sites. Coumarin ring acts by different mechanisms on the cancer cells and is also known for induction of apoptosis. These hybrid compounds showed a good anticancer profile with lower IC50 values on cell lines used to study. In cell cycle analysis, hybrids reportedly arrest the G2-M-phase and were found to induce the apoptosis too (Belluti et al., 2010).
The retinoid-chalcone hybrids
In these hybrids, two potent anticancer molecular entities that is retinoid moiety of bexarotene and chalcone moiety (4) are fused together. The hybrids show anticancer effect in low micromolar range against colon adenocarcinoma HT-29 cell line (Mizuno et al., 2010).
 | [Click here to view] |
Strategy II (Making Combi-molecules of two or more different drugs)
The second approach deals with a combination of two or more drugs with the help of linker or spacers. The compounds of this category are also recognized as combi-molecules and the connection can be done via a cleavable or non-cleavable linkage. The perspective of non-cleavable linkage is based on the ability of the different molecules to retain their biological properties and efficacy up to they reach to their biological targets. On the other side, the approach of cleavable bond is based on the release of either two active molecules or one active molecule under physiological conditions that prevail at the site of action. To improve poor pharmacokinetic properties of the anticancer drugs or for the slowly deliver of therapeutic entities generally ester, amide or carbamate linkages are incorporated among the molecules. Moreover, to improve the selectivity and specificity toward cancer cells, produg approach is also in a fashion that releases two active drugs directly at the targeted sites (e.g., phosphorylated diethylstilbestrol (DES) prodrugs for prostate cancer) (Fortin and Bérubé, 2013).
This combination strategy further classified into three sub-strategies, which are as follows:
Combining the two drugs with the same mechanism of action
In this, moieties having the same mechanism of action are fused together to make combi-molecules so that efficacy and affinity toward the common target are increased. The molecules are fused in such a way that the site of action of the drug is not affected but at last, the hybrid obtained is versatile. Examples are:
 | [Click here to view] |
Pyrazoline-coumarin hybrids
These hybrids were prepared by linking the pyrazolin-5-one heterocyclic ring with coumarin moiety using an acetoxy group as joiner (5), with the aim to have geiparvarin (a coumarin derivative) like anticancer activity on various cancer cell lines. These hybrids were found to show moderate anticancer activity on different cancer cell lines (Ismail et al., 2010).
1H-1,2,3-triazole tethered Isatin hybrids
They were synthesized by combining two Isatin moieties using 1,2,3-triazole linker in-between (6). The most active hybrids depicted their antiproliferative action in the nanomolar range on different cell lines (Singh et al., 2012).
Combining the two drugs with the different mechanisms of action
These combi-molecules are prepared to counteract multi-biological targets. The purpose of this strategy is to obtain a potent, effective, and useful therapeutic hybrid. This is a synergistic approach to tumor cell via different mechanisms. Examples of these hybrids are as follows:
Nitric oxide (NO) releasing chalcone hybrid
Anticancer activity in tumor cell is found to be mediated via. NO release and it may prevent metastasis also. In view of this, NO donating--chalcone hybrids (7) were prepared by tethering NO-releasing nitrate ester (ONO2) over the moiety of chalcone. Results depicted that chalcone hybrids with NO releasing group effectively inhibit the cancer cells proliferation when studied over various cell line panels (Mourad et al., 2012).
Pyrrolobenzodiazepine –acridone hybrid
Acridones have been reported to possess Deoxyribonucleic acid (DNA) intercalation property and show potent anticancer activity. Conjugates of pyrrolobenzodiazepine and acridone (8) exhibit average lethal dose (LC50) in the nanomolar range for different cancer cell lines and also reported to have DNA-binding property too (Kamal et al., 2004).
 | [Click here to view] |
Combining drugs that target specific biological tissues
This combi-molecular strategy is used to synthesize novel anticancer hybrids of two different drugs that specifically target on affected tissues. Hybrids prepared using this approach are highly specific, efficacious, and less toxic. Examples of these combi-molecules are as follows:
Nitrogen mustard and tyrosine hybrid
To mimic the same action as of estradiol nucleus, combi-molecules of aromatic nitrogen mustards and tyrosine (9) were designed and synthesized. Antiproliferative assay exhibits that hybrids inhibit the multiplication of prostate, breast, ovarian, and uterine cancer cell lines in micromolar range and also more active than chlorambucil drug (Descôteaux et al., 2012).
Nitroxyl-aziridine hybrid
Aziridine is an important scaffold, which is reported to have a good anticancer profile. To further increase the effectiveness, novel hybrid was designed by linking the aziridinyl to nitroxyl moiety (10). The hybrids are reported to possess activity against mice melanotic melanomas (type of skin cancer) (Kumamoto et al., 2010).
 | [Click here to view] |
All the above-mentioned contents show the developments that are going on and the hybrid molecules are continuously being produced via different strategies to act as potential anticancer agents. Thus, the hybrid approach is now become a fashion to develop those potent drugs which nobody has ever thought about. It is mainly expected that hopefully in future, hybrid drugs will definitely decrease the mortality and morbidity rate manifolds.
Uracil and 5-FU
Uracil (pyrimidine 2,4 dione) is a common nitrogen containing nucleobases present in the nucleic acid of Ribonucleic acid (RNA). During RNA sequencing, Uracil makes two hydrogen bonds with Adenine, whereas in DNA sequencing the thymine replaces Uracil base. In laboratory, Uracil can be easily prepared by the following methods mentioned in Figure 4
Uracil undergoes tautomeric shifts and gives rise to amide-imidic acid tautomers. These tautomers are maximum formed and stable at pH 7. The common form of Uracil is amide lactum tautomer.
 | [Click here to view] |
 | Figure 3. Hybrid strategy by making combi-molecules. [Click here to view] |
 | Figure 4. Basic synthetic schemes for Uracil. [Click here to view] |
Concerning to drug designing, Uracil has been considered as a versatile moiety that imparts drug like properties to the designed molecules. Since years, to explore chemotherapeutic potential, various Uracil derivatives, hybrids, metal complexes, and prodrugs are continuously being prepared by making substitutions at N1, N3, C5, and C6 positions (Dimitrova et al., 2017; Newkome and Pandler, 1982). As of now several Uracil derivatives or Uracil fused hybrids are well known for their antiviral, antibacterial, and antitumor potential (Pałasz and Ciez, 2015). Due to their DNA intercalation properties, Uracil derivatives and 5-FU interfere in various metabolic pathways of cell division and confer the cytotoxic potential. Both 5-FU and 5-chlorouracil (Fig. 6) were the first Uracil derivatives that showed therapeutic potential (Abdel-Mottaleb and Abdel-Mottaleb, 2016). Among them, 5-FU has had clinical success as a single agent and became a useful drug to treat solid tumors like colon, breast, and other cancers. But 5-FU has not gained much importance due to various limitation associated with it, like poor selectivity, quick drug resistance, undesired side effects, fast catabolism, wide distribution, and short half-life (Fata et al., 1999; Zhang et al., 2008).
So as to reduce the toxicity, side effects, and also to reverse the acquired resistance, several structural modifications have been made by make derivatives, prodrugs, nucleosides, metal complexes, and hybrids of Uracil and 5-FU both. But the present article mainly portrays the earlier and recently reported hybrids of Uracil or 5-FU that have impressive anticancer potential.
Hybrids of Uracil and 5-FU
Since from the last two decades, scientists are continuously putting their efforts in making hybrids of Uracil and 5-FU by linking them with other bioactive pharmacophores. Joining of Uracil moiety with other molecule was found to be fruitful especially in the case of anticancer profile. Table 1 contains a list of Uracil/5-FU hybrids synthesized and reported before the year 2014. The table also depicts a short description of anticancer activity of each hybrid.
 | Figure 5. Tautomers of Uracil. [Click here to view] |
 | Figure 6. Firstly reported Uracil derivatives (having therapeutic potential). [Click here to view] |
Earlier reported (Before the year 2014) hybrids of Uracil or 5-Flourouracil
 | Table 1. Highlights of earlier reported Uracil/5-FU hybrids with their anti-cancer profile [Click here to view] |
Recently developed anticancer Uracil/5-FU hybrids
The successful efforts of the scientists in the field of hybrids have kept the interest of many medicinal chemists ongoing. Being impressed by the results of previous researches, in past years, scientists have designed and synthesized some more Uracil/5-FU hybrids and were evaluated for their anticancer profile, which again has shown promising results on different kinds of cancer. Figure 7 displays the novel combinations, which were made after the year 2014 (last five years). Moreover, the detailed description of individual hybrid like their synthetic approach, cancer targets, IC50 values of potent hybrids on panel of cancer cell lines used, and other outcome of the research is mentioned below.
5-Fluorouracil-cholesterol hybrid
Tumor cells engulf more low density lipoprotein (LDL) as compared to normal cells due to over expression of LDL receptors (LDLR) on cancerous cells (Gueddari et al., 1993; Yen et al., 1995). Keeping a view on elevated level of LDLR on cancer cells, Awwad A. Radwan et al. designed and synthesized novel 5-FU-Cholesterol conjugates expecting to be rapidly taken up by LDLR, which subsequently leads to internalization into cancer cells via LDL-receptor-mediated channels. Synthetic approach of desired hybrids (Scheme 1) starts with refluxing cholesterol (12) with dicarboxylic acid anhydride (11) in the presence of pyridine yield the compounds (13-15), which on further agitation with 5-FU in the presence of DCC, 4-dimethylamino-pyridine (DMAP) using dimethyl formamide (DMF), Tetrahydrofuran (THF) (1:1 v/v) solvent at 50°C for 2 days to get the target hybrids (16-18).
Cytotoxic potential of all the hybrids was evaluated against breast cancer cells (MDA-MB-231) and lovo colon cells grew in culture media. Amid all compound 16 (-X-L- = -CH2CH2-) was found to be the most potent at 100 μM concentration. Moreover, in-vivo anticancer activity was also performed using mice model of Solid Ehrlich Carcinoma (SEC). Results showed that 5FU-cholesterol hybrid 16 produced a significant decrease in tumor volume of SEC on dosing for 3 mg·Kg−1 as compared to the 5-FU (Radwan and Alanazi, 2014). This research represents hybrid 16 could be utilized as a prodrug in future for target delivery of 5-FU via. LDLR mediated pathway into tumor cells.
5-Fluorouracil-colchicine hybrids
Colchicine derivatives have been reported to possess tumor suppression ability and are also regarded as mitotic poisons (Bhattacharyya et al., 2008). Based on some structure-activity relationship (SAR) of colchicines, it was clarified that C-10 analogues of colchicine possess better efficacy and lower toxicity than Colchicine itself (Kozaka et al., 2010). Following this concern, Shen et al. (2015) synthesized a library of novel hybrids having C-10 derivatives of Colchicine with 5-Fluorouracil-1-yl-acetic acid linked through a coupling reaction. Synthetic approach for the most potent hybrid is depicted in Scheme 2. In the first step N1 derivative 20 was easily synthesized by treating 5-FU (19) with chlorooacetic acid in 10% KOH solution at room temperature for 2 hours. Colchicine (21) was initially converted to thiocolchicine (22) in the presence of NaSCH3, and then N-deacetylthiocolchicine compound 23 was obtained by refluxing 22 with 2 mol/L HCl in CH3OH. The target hybrid compound 24 was obtained by acylation of the compound 23 in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and DMAP in dry CH2Cl2 at room temperature.
 | Figure 7. Latest hybrids of Uracil/5-Fluorouracil linked with different pharmacophores. [Click here to view] |
In-vitro anticancer potential was tested against liver cancer cells (BEL7402), ovarian cancer cells (A2780), lung adenocarcinoma cells (A549), and breast cancer cells (MCF-7) (Shen et al., 2015). The bioassay results depicted that some compounds possess significant activity, but compound 24 was found to be the most effective against all four cell lines with least IC50 values (A2780 = 9.5 μM, BEL7402 = 10.2 μM, A549 = 7.8 μM, MCF-7 = 7.5 μM).
5-FU-deoxypodophyllotoxin hybrids
Podophyllotoxin (PPT) is a naturally occurring cyclolignan, which have been reported to possess potent cytotoxic effects on different cancer cell lines (Canel et al., 2000). The structural analogue of PPT that is 4-deoxypodophyllotoxin (DPT) is also proved to exhibit antiproliferative and antitumor activity in several cell types (Xiao et al., 2018). Further in in-vivo studies, 4′-demethyl-4-deoxypodophyllotoxin (DDPT) derived from DPT was found to be equipotent to DPT (Huang et al., 2012). Based on the idea of synergistic effect, Guan et al. (2016) prepared compound hybrid series of DDPT and 5-FU using various diamine linkers. Formation of most potent hybrids involves two step syntheses. In step-I, compound 20 was prepared by reacting 5-FU (19) with chloroacetic acid in 10% KOH solution. Then compound 20 was reacted with various N-Boc diamines using 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI), N-hydroxybenzotriazole (HOBt) in DMF to yield compounds 25. Afterwards, compounds of 25 were deprotected using trifluoroacetic acid in dichloromethane to give compounds 26. In step II, DDPT (27) is reacted with succinic anhydride under DMAP and Triethylamine (TEA) in dichloromethane to obtain 28. Eventually, 28 reacted with 26 in the presence of HOBt and EDC to yield desired hybrid 29.
All the synthesized hybrids were evaluated against four different cancer cell lines (liver cancer cells HEPG2, lung cancer cells A549, cervical cancer cells HeLa, and colorectal adeno-cancer cells HCT-8) and one healthy cell line (human lung fibroblast WI-38), using etoposide, DDPT and 5-FU as standard compounds. Among them hybrid compound 29 (n = 4) is significantly potent against all cell lines with IC50 values of 4.03, 0.27, 1.01, and 0.45 μM in HeLa, A549, HCT-8, and HEPG2 cells, respectively. Moreover, compound 29 proved to be less toxic in comparison to 5-FU and etoposide against normal human lung fibroblast cells WI-38. Flow cytometry analysis in A549 cells depicted that compound 29 caused the cell-cycle arrest in the G2/M phase (Guan et al., 2016). Concluding the above study suggests that compound 29 can serve as a novel anticancer drug candidate in future.
Similarly, Xiang et al. (2017) have synthesized numeral hybrid compounds of deoxypodophyllotoxin and 5-FU using various linkers targeting Matrix metalloproteases (MMPs) (Huang et al., 2012). MMPs present in epithelial cells hails from the group of Zinc-dependent proteases that can specifically digest some extracellular matrix components. Some MMPs specially MMP-2 and MMP-9 reportedly participate in initiating and switching angiogenesis (Stetler-Stevenson, 1999; Van Hinsbergh and Koolwijk, 2008). The β-catenin signaling pathway regulates various processes like cell proliferation, differentiation, apoptosis, and migration (Prakash and Swaminathan, 2015). Any mutation in β-catenin signaling evokes diverse range of cancers (Clevers and Nusse, 2012).
 | Scheme 1. Synthetic strategy of 5-FU-cholesterol hybrids [Click here to view] |
Among all synthesized hybrids, compound 30 was found to possess cytotoxic properties for cancerous cells and least toxic toward non-cancerous cells in comparison to standard Etoposide. In further studies, at a very low concentration (0.1–0.3 μM) compound 30 is found to inhibit proliferation, metastasis, tube formation, and disarray the formed tube-like structures of human umbilical vein endothelial cells. Results also depicted that compound 30 inhibits angiogenesis by decreasing the level of MMP-2 and MMP-9 both (Xiang et al., 2017).
 | [Click here to view] |
Ferrocenyl containing Uracil hybrids
Uracil-metallocene hybrids
Metallocene-nucleobase conjugates have become important part of bio-organometallic chemistry. Bi-conjugates of ferrocenyl-nucleobase have already been reported to show good anticancer activity against breast cancer cells MCF-7 and colon cancer cells HT-29 (Kowalski et al., 2015). With the aim to increase anticancer potential by increasing Ferrocenyl nuclear systems, Skiba et al. (2015) synthesized novel metallic mono-, di-, and tri-nuclear Ferrocenyl-Uracil derivatives. All conjugates are having a linker (-CH2CH2CH2-) bridge between Ferrocenyl and Uracil end-groups.
Synthetic approach toward these hybrids is mentioned in Scheme 4. Reaction initiated by reacting (3-chloropropionyl)-metallocenes (30, 31, 32) with 5-methyl Uracil to give Michael adducts (33, 34, 35). Afterwards, adducts were treated with 5-ethyl-2-methylpyridine borane (PEMB) in acetic acid for 2 hours at room temperature which yields desired hybrids (36, 37, 38) containing trimethylene (-CH2CH2CH2-) linker.
Anticancer screening depicts that di-nuclear hybrid 38 was found to be potent against HT-29 colon carcinoma cells with less IC50 value 4.3 ± 0.7 μM compared to standard Cisplatin (IC50 = 7.5 ± 0.4 μM). Moreover, hybrid 38 (IC50 = 2.4 ± 0.2) produced similar action as standard Cisplatin (IC50 = 2.9 ± 1.0μM) against non-tumor mouse L929 fibroblast cells. Research also showed that incorporation of heavy and lipophilic Fc-CH2-Fc-group enhanced the cytotoxic effects and also the presence of a single metallocenyl moiety led to inactive conjugates (Skiba et al., 2015). Hence, the dinuclear systems conjugated with organometallic compounds were found to be beneficial for anticancer agents.
Uracil-ferrocenylchalcone hybrids
Ferrocenyl conjugates with other moieties have been proved to be potent anticancer agents (Tan et al., 2012). 5-FU, an antimetabolite drug commonly used for the treatment of cancers like colorectal, stomach, and breast cancer. However, therapeutic importance of 5-FU is decreased due to its poor selectivity, Central Nervous System toxicity and other associated side effects (Pan et al., 2011). Chalcones too possess the cytotoxic potential as they exhibit the same mode of action as Combretastatins (Pettit et al., 2005). This collective information prompted Singh et al. (2018) to synthesize a series of 1,2,3 triazole tethered hybrids of Uracil and Ferrocenylchalcone via. azide alkyne cycloaddition reaction. Synthetic route of the most potent hybrids is shown in Scheme 5.
 | Scheme 2. Synthetic strategy of 5-FU-colchicine hybrids [Click here to view] |
Synthetic methodology involves the hydride promoted alkylation of 5-substituted Uracils (39) using dibromoalkanes to yield derivatives 40. Then N-alkylazido5-substituted Uracil derivatives (41) were prepared by reacting 40 with sodium azide (NaN3) in DMF. Finally, 1H-1,2,3-triazole tethered uracil–ferrocenyl chalcone hybrids 43a-g were prepared using Cu-catalyzed azide-alkyne cycloaddition reaction between N-alkyl azido-uracils (41) and O-propargylated ferrocenyl-chalcone (42). Prepared hybrids were further evaluated for their cytotoxic profile on human leukemia cell lines (CCRFCEM) and human breast adenocarcinomacell lines (MDA-MB-468) using Doxorubicin as standard drug. The results depicted that several hybrid compounds 43a-g which are having longer chain spacer (n = 5,6,8) were found to inhibit the cell proliferation up to 70% after 72 hours of 50 μM dosing. Potential compounds with longer chain spacer were also evaluated for the cytotoxic profile against normal kidney cell lines (LLC-PK1 ATCC CL-101) and all were found to be non-toxic toward healthy cells, even after 72 hours (Singh et al., 2018).
Uracil N1 monosubstituted and N1, N3 bisubstituted 1,2,3 traizole hybrids
Gregorić et al. (2017) synthesized two novel series of hybrid compounds of pyrimidine 2, 4 dione (Uracil). First series was of 1,4-disubstituted 1,2,3-triazole tethered pyrimidine-2,4-dione hybrids (47a-c) and second was 6-alkylethynylfuro[2,3-d]pyrimidine-2-one–1,2,3-triazole hybrids (48).
The synthetic routes of both the series are mentioned in Scheme 6, reaction initiated with propagylation of 5-bromo uracil (44) that yields N1-monosubstituted (45) and N1,N3-bisubstituted (46) prop-2-ynyl derivatives. First series of compounds that is N1,N3-bisubstituted 1,2,3-triazole hybrids of 5-bromouracil (47a-c) was prepared by copper supported azide-alkyne cyclization of 46 by sodium azide (NaN3) in the presence of sodium ascorbate using solvent DMF. Second line of compounds was prepared by coupling 1,2,3-triazole–5-bromo uracil hybrids (48) with terminal alkynes including cyclopropylethyne, 5-chloropent-1-yne, 1-ethynyl-4-pentylbenzene, and oct-1-yne and the presence of palladium catalyst Tetra-kis (triphenylphosphine) palladium(0) [Pd(PPh3)4], N,N-diisopropylethylamine (N,N-DIPEA) and Cu(I) iodide to get the target hybrids 49.
Both the synthesized library of hybrids were further evaluated for their antiproliferative potential on five different cancerous cell lines that are lung cancer cell lines (A549), liver cancer cell lines (HEPG2), ductal pancreatic cancer cell lines (CFPAC-1), cervical carcinoma cell lines (HeLa), and metastatic colorectal cancer cell lines (SW620). Among the first series N,N-1,3-bis-(1,2,3-triazole)-5-bromouracil hybrids 47a-c were found to show the best inhibition toward all cell lines with least IC50 value (Figure 8).
In the second series, 5,6-disubstituted fluoro [2,3-d]pyrimidine-2-one-1,2,3-triazole hybrid 49a exhibits the most potent cytostatic effect especially against HEPG2 and HeLa cell lines as compared to the standard 5-FU (Gregorić et al., 2017).
 | [Click here to view] |
IC50 (49a) = 2.67μM (HEP-G2), 6.51 μM (HeLa)
IC50 (5-FU) = 8.90μM (HEP-G2), 8.81 μM (HeLa)
Uracil-ubenimex hybrid
CD-13 is a multifunctional protein that is abundantly expressed on various cell surfaces. CD-13 is reported to play an important role in cancer invasion (Saiki et al., 1993), migration, angiogenesis, and resistance to apoptosis. Ubenimex is the solely marketed CD-13 inhibitor and immunomodulator in the treatment of leukemia (Ota et al., 1986). Moreover, it is used complimentary with other potential agents in the treatment of cancer (Ota and Uzuka, 1992). Combination therapy of cytotoxic agent 5-FU with other moieties is already proved to reduce the tumor volume incredibly as compared to a single drug (Li et al., 2015). All reported facts stimulated Yuqi Jiang and team to synthesize a novel hybrid prodrug 57, which was supposed to release 5-FU and Ubenimex in-vivo. In hybrid molecule 57, both the potential candidates 5-FU and Ubenimex are connected via. –O—CH2— linker. The synthetic reaction initiated with the formation of 50 from 19 (5-FU) in the presence of 37% oxymethylene. The reaction of 50 with (S)-2-((tert-butoxycarbonyl)-amino)-4-methylpentanoic acid (51) in the presence of 1, 3-dicyclohexyl-carbodiimide (DCC) and DMAP yields acid amide 52. Then deprotection of 52 gives the product 53. On the other side, compound (3S, 4R)-4-amino-3-hydroxy-2-oxo-5-phenylpent-anoic acid (54, AHPA) was protected by Boc (ditert-butyl dicarbonate) group to produce 55. Then intermediate 56 was produced by reacting 55 with 53 in the presence of EDCI and 1- HOBt. Then on the treatment of 56 with hydrogen chloride (HCl) in acetidin yields the target hybrid 57.
In-vivo and in-vitro evaluation study depicted that hybrid 57 moderately inhibits CD-13 and found to produce better antiproliferative, antimetastasis, and antiangiogenesis effects than standard 5-FU and Ubenimex. Hybrid 57 shows the best results against A-549 lung cancer cell lines with IC50 value 5.68 μM in comparison to standard 5-FU with IC50 = 16.96 μM and Ubenimex with IC50 = 16.29 μM. Even in 5-FU resistant mice model, hybrid 57 proved more efficient as compared to 5-FU or 5-FU plus Ubenimex (Jiang et al., 2016).
Continuing the same research, Jiang et al. further prepared a novel series of derivatives of hybrid 57 using various linkers to prepare a stable conjugate of 5-FU-ubenimex. Compound 60a-e were prepared from 5-FU (19) by using various Boc protected amino acids and followed by their deprotection as mentioned in Scheme 7. On the other hand, compound 54 was condensed with L-leucine benzyl ester toluene-4-sulfonate and simultaneously Boc protected to form 61, which was further protected by 2,3 dihydropyran and followed by deprotection to give the acid 62. Reaction of 62 with 60-a-e in the presence of EDCI and HOBt gave the intermediates 63a-e. At the end, Boc deprotection of compounds 63a-e with HCl in acetidin yields the desired hybrids 64a-e.
 | Scheme 3. Synthetic strategy of 5-FU-deoxypodophyllotoxin hybrids [Click here to view] |
Amidst all compounds hybrid 64a was found to possess the most potent in-vitro antiproliferative action with IC50 = 10–26 μM, pro-apoptosis, antimetastasis, antiangiogenesis, and CD13 inhibition (IC50 = 0.18–5.69 μM) too. Stability and pharmacokinetic study depicted that hybrid 64a released Ubenimex and 5-FU slowly at the desired location. Hence, it was proven to be a good mutual hybrid prodrug of 5-FU and Ubenimex. Also, hybrid 64a also showed superior growth inhibition in 5-FU-resistant mice model with liver cancer (Jiang et al., 2018).
Uracil-camptothecin hybrids
As Camptothecin has been reported as an important class of antitumor agents (Wall et al., 1966), Di-Zao Li et al. synthesized series of novel ester linkage conjugates of Uracil & Camptothecin. Uracil C5-substituted derivatives (65a-e) were derivatized using chloroacetic acid in the presence of KOH in water. Further N1 substituted derivatives (66a-e) were condensed with 7-ethyl camptothecine (67) in the presence of EDCI, DMAP, and pyridine to yield the target hybrids (68a-e).
 | Scheme 4. Synthetic scheme of Uracil-metallocene hybrids [Click here to view] |
 | Scheme 5. Synthetic strategy of Uracil-ferrocenylchalcone hybrids [Click here to view] |
All the novel conjugates were tested for antitumor activity against five different cancer cell lines (A549, BEL7402, BGC-823, HCT-8, and A2780). These results depicted that the hybrid 68b (IC50 = 0.007–0.147 μM) & 68c (IC50 = 0.002–0.670 μM) possess the best efficacy against all cancer cell panel in comparison to individual Camptothecin and Topotecan (standard). Furthermore, in-vivo evaluation of compound 68b & 68c (Fig. 9) for anticancer activity against mice liver carcinoma H22 depicted that they have a nearly similar response as of Paclitaxel and Cyclophosphamide.
 | Scheme 6. Synthetic strategy of Uracil N1 monosubstituted and N1, N3 bisubstituted 1,2,3 traizole hybrids [Click here to view] |
 | Figure 8. 1, 2, 3-Tethered Uracil Hybrids with their IC50 value. [Click here to view] |
Similar to Camptothecin, hybrid 68b was also found to inhibit topoisomerase-1 more efficiently (Li et al., 2017). Hence, hybrid 68b with high potency and lower toxicity can be developed as a potential drug candidate in future.
Uracil-oleanolic acid hybrid
Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid) belongs to the family of oleanane pentacyclic triterpenes, which is reported to be isolated from more than 1620 different plant species of food and herbs (Pollier and Goossens, 2012). Oleanolic acid analogues have been proved to have different biological activities including antiproliferative activity in solid tumor cells. SAR studies of oleanolic acid showed that structural modification at C-3 and C-28 improves cytotoxic properties against prostate cancer (PC-3), lung cancer (A549), and breast cancer (MCF-7) cell lines (Hao et al., 2013). Being inspired by the evidence, Mo et al. (2016) synthesized the conjugates of Acyl-oleanolic acid and Uracil. Synthetic pathway starts with the treatment of oleanolic acid (69) (1 equiv) with anhydride (1.5 equiv) and DMAP (0.1 equiv) in anhydrous CH2Cl2/pyridine at room temperature to yield 3-O-acyl derivatives (70a-c). These derivatives were then initially treated with oxalyl chloride (18 equiv) to give the corresponding acyl chloride, which was further treated with Uracil (3 equiv) in the presence of Et3N (triethyl amine) to produce final acyl oleanolic acid-uracil hybrids (71a-c).
 | Scheme 7. Synthetic approach toward 5-FU-ubenimex hybrids [Click here to view] |
 | Figure 9. Most potent Uracil-Camptothecin hybrids. [Click here to view] |
All synthesized hybrids were evaluated against five cancer cell lines (HEP-G2, A549, BGC-823, MCF-7, and PC-3) for cytotoxic potential taking Oleanolic acid and 5-FU as standard. Almost all the conjugates proved to be cytotoxic with IC50 value of less than 10 μM. Among them, compound 71c (mentioned below) was found to be the most effective against all cell lines with IC50 ranging from 0.22 to 6.99 μM.
Moreover, unlike other compounds, compound 71c was found to be the least cytotoxic to healthy liver cell line (HL-7702). Further cell cycle analysis depicted that compound 71c could trigger apoptosis by activating caspase-3/9 and arrest the G1 phase of HEP-G2 cells (Mo et al., 2016).
 | Figure 10. SAR of Uracil with respect to hybridization. [Click here to view] |
Structure activity relationship (uracil hybrids)
On the thorough study of earlier and recently reported Uracil and 5-FU hybrids, it can be summarized via basic Uracil moiety (Fig. 10) that:
- N1 and N3 are the foremost hybridization sites for the generation of mono and bifunctional hybrids.
- In order to provide the rigidity, other pharmacophores linked directly to N1 and N3 through ester or amide linkage (Uracil-camptothecine hybrid) and flexible hybrids are obtained by linking Uracil with other pharmacophores via a chain linker (Uracil-Ferrocenylchalcone hybrids, 5FU- deoxypodophyllotoxin hybrids, etc.).
- Oxo groups at second and fourth positions usually participate in h-bond formation with amino acid residues of target protein. Replacement of “=O” (oxo) with “=S” (thio group) produces another class of anticancer agents that is thiouracils (El-Naggar et al., 2017). Sometime oxo groups are utilized in making fused hybrids also (furan fused triazole linked hybrids, 49a)
- Use of long-chain linker at N1, N3 and alkyl group substitution at C6 yield hybrid having good cellular penetration power.
- Halogen substitution at C5 position increases the cytotoxic potential. Fluorine produces the maximum inhibition and the order is F>Cl>Br>I>H. Inhibitory potential of 5-FU is reported to be increased via hybridization with other pharmacophores (5FU-Cochicine hybrids, 5FU-cholesterol hybrids, 5FU-PPT hybrids, etc.).
- C5–C6 double bond is the best site for making adduct via Diels-Alder reaction Or making fused hybrids with other moieties (Uracil-bischalcone hybrids).
 | Scheme 8. Synthetic approach toward 5-FU-Ubenimex hybrids (using various linkers) [Click here to view] |
 | Scheme 9. Synthetic scheme of uracil-camptothecin hybrids [Click here to view] |
 | Scheme 10. Synthetic scheme of uracil-oleanolic acid hybrids [Click here to view] |
Hence, the present study proves the versatility of Uracil moiety, which encourages the medicinal chemistry for its consideration in drug designing strategies with other pharmacophores to produce multi-targeted hybrids.
CONCLUSION
From several years, continuous efforts are being made for increasing the efficacy and target specificity of 5-FU and other Uracil derivatives by making their prodrugs, hybrids, conjugates, metal-ligand complexes, etc. Among them, in cancer treatment, the hybrid approach remains one of the best strategies to overcome the limitations associated with single drug component and multiple dosing of drugs. This prompted us to collectively depict the anticancer potential of recently developed Uracil and 5-FU hybrids. Based on the outcomes of all the hybrids in concern to their IC50 values and other pharmacological results, it can be positively concluded that making hybrids of Uracil and 5-FU are found to be beneficial in terms of cancer cell specificity, multiple drug resistance, target drug delivery, drug toxicity, and dose reduction. Thus, this review study suggests that there is still a significant scope in developing novel anticancer hybrid drugs by placing an active pharmacophore at different positions of Uracil nucleus. Hopefully, the continuous efforts of medicinal chemists of hybridization of Uracil or 5-FU with a diversity of potential molecules will probably discover a magical anticancer hybrid, which will be proven as a boon in future.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
The author declared that there is no conflict of interest.
ABBREVIATIONS
AHPA (3S, 4R)-4-amino-3-hydroxy-2-oxo-5-phenyl‑pent-anoic acid
DCC N,N′-Dicyclohexylcarbodiimide
DCM Dichloromethane
IC50 values half maximal inhibitory concentration
N,N-DIPEA N,N-diisopropylethylamine
PEMB 5-ethyl-2-methylpyridine borane
PPTS Pyridinium p-toluenesulfonate
TEA Triethylamine
TFA Trifluoroacetic acid
THF Tetrahydrofuran
REFERENCES
Abdel-Mottaleb Y, Abdel-Mottaleb MSA. Molecular modeling studies of some uracil and new deoxyuridine derivatives. J Chem, 2016; 2016:1–12. CrossRef
Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. J Control Release, 2011; 153(3):198–205; doi:10.1016/j.jconrel.2011.06.001 CrossRef
Baraldi PG, Romagnoli R, Guadix AE, Pineda de las Infantas MJ, Gallo MA, Espinosa A, Martinez A, Bingham JP, Hartley JA. Design, synthesis, and biological activity of hybrid compounds between uramustine and DNA minor groove binder distamycin A. J Med Chem, 2002; 45(17):3630–8. CrossRef
Belluti F, Fontana G, Bo LD, Carenini N, Giommarelli C, Zunino F. Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: identification of novel proapoptotic agents. Bioorg Med Chem, 2010; 18(10):3543–50. CrossRef
Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev, 2008; 28(1):155–83. CrossRef
Cai TB, Tang X, Nagorski J, Brauschweiger PG, Wang PG. Synthesis and cytotoxicity of 5-fluorouracil/diazeniumdiolate conjugates. Bioorganic Med Chem, 2003; 11(23):4971–5. CrossRef
Cancer Statistics. Case RAM. Br Med J [Internet], 2007; 2(5466):866. Available from http://www.jstor.org/stable/25404010 (Accessed 24 September 2019).
Canel C, Moraes RM, Dayan FE, Ferreira D. Podophyllotoxin, Phytochemistry, 2000; 54(2):115–20. CrossRef
Chen SW, Xiang R, Liu J, Tian X. Synthesis and biological evaluation of novel conjugates of podophyllotoxin and 5-FU as antineoplastic agents. Bioorganic Med Chem, 2009; 17(8):3111–7; doi:10.1016/j.bmc.2009.03.009 CrossRef
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell, 2012; 149(6):1192–205. CrossRef
Descôteaux C, Brasseur K, Leblanc V, Parent S, Asselin É, Bérubé G. Design of novel tyrosine-nitrogen mustard hybrid molecules active against uterine, ovarian and breast cancer cell lines. Steroids, 2012; 77(5):403–12. CrossRef
Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, Jacks T. Public access NIH public access. PLoS One, 2017; 32(7):736–40.
El-Naggar AM, Abou-El-Regal MM, El-Metwally SA, Sherbiny FF, Eissa IH. Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol Divers, 2017; 21(4):967–83. CrossRef
Fata F, Ron IG, Kemeny N, O’Reilly E, Klimstra D, Kelsen DP. 5-Fluorouracilinduced small bowel toxicity in patients with colorectal carcinoma. Cancer, 1999; 86:1129–34. CrossRef
Fortin S, Bérubé G. Advances in the development of hybrid anticancer drugs. Expert Opin Drug Discov, 2013; 8(8):1029–47. CrossRef
Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker BL, Stewart L, Cushman M. Design, synthesis, and biological evaluation of cytotoxic11-alkenylindenoisoquinoline topoisomerase I inhibitors and indenoisoquinoline-camptothecin hybrids. J Med Chem, 2003; 46(15):3275–82. CrossRef
Gregorić T, Sedić M, GrbÄić P, Tomljenović Paravić A, Kraljević Pavelić S, Cetina M, Vianello R, Raić-Malić S. Novel pyrimidine-2,4-dione–1,2,3-triazole and furo[2,3-d]pyrimidine-2-one–1,2,3-triazole hybrids as potential anti-cancer agents: synthesis, computational and X-ray analysis and biological evaluation. Eur J Med Chem, 2017; 125:1247–67. CrossRef
Guan XW, Xu XH, Feng SL, Tang ZB, Chen SW, Hui L. Synthesis of hybrid 4-deoxypodophyllotoxin-5-fluorouracil compounds that inhibit cellular migration and induce cell cycle arrest. Bioorganic Med Chem Lett, 2016; 26(6):1561–6. CrossRef
Gueddari N, FavreG, Hachem H, Marek E, le Gaillard F, Soula G. Evidence for up-regulated low density lipoprotein receptor in human lung adenocarcinoma cell line A549. Biochimie, 1993; 75:811–9. CrossRef
Hao J, Liu J, Wen X, Sun H. Synthesis and cytotoxicity evaluation of oleanolic acid derivatives. Bioorg Med Chem Lett, 2013; 23(7):2074–7. CrossRef
Huang WT, Liu J, Liu JF, Hui L, Ding YL, Chen SW, Synthesis and biological evaluation of conjugates of deoxypodophyllotoxin and 5-FU as inducer of caspase-3 and -7, Eur J Med Chem, 2012; 49:48–54. CrossRef
Ismail MMF, Rateb HS, Hussein MMM. Synthesis and docking studies of novel benzopyran-2-ones with anticancer activity. Eur J Med Chem, 2010; 45(9):3950–9. CrossRef
Jiang Y, Li X, Hou J, Huang Y, Jia Y, Zou M, Zhang J, Wang X, Xu W, Zhang Y. Discovery of BC-01, a novel mutual prodrug (hybrid drug) of ubenimex and fluorouracil as anticancer agent. Eur J Med Chem, 2016; 121:649–57. CrossRef
Jiang Y, Li X, Hou J, Huang Y, Wang X, Jia Y, Wang Q, Xu W, Zhang J, Zhang Y. Synthesis and biological characterization of ubenimex-fluorouracil conjugates for anti-cancer therapy. Eur J Med Chem, 2018; 143:334–47. CrossRef
Kamal A, Srinivas O, Ramulu P, Ramesh G, Kumar PP. Synthesis of C8-linked pyrrolo[2,1-c] [1,4]benzodiazepine-acridone/acridine hybrids as potential DNA-binding agents. Bioorg Med Chem Lett, 2004; 14(15):4107–11. CrossRef
Kowalski K, Skiba J, Oehninger L, Ott I, Solecka J, Rajnisz A, Therrien B. Synthesis and anticancer activity studies of ferrocenyl-thymine-3,6-dihydro-2H-thiopyranes - a new class of metallocene-nucleobase derivatives. J Organomet Chem, 2015; 794:216–22. CrossRef
Kozaka T, Nakagawa-Goto K, Shi Q, Lai CY, Hamel E, Bastow KF, Brossi A, Lee KH. Antitumor agents 273. Design and synthesis of N-alkyl-thiocolchicinoids as potential antitumor agents. Bioorg Med Chem Lett, 2010; 20(14):4091–4. CrossRef
Kucuksayan E, Ozben T. Hybrid compounds as multitarget directed anticancer agents. Curr Top Med Chem, 2017; 17(8):907–18. CrossRef
Kumamoto T, Suzuki K, Kim SK, Hoshino K, Takahashi M, Sato H, Iwata H, Ueno K, Fukuzumi M, Ishikawa T. Synthesis, physical properties, and cytotoxicity of Nitroxyl-Aziridine hybrid. Helv Chim Acta, 2010; 93(11):2109–14. CrossRef
Kumar K, Sagar S, Esau L, Kaur M, Kumar V. Synthesis of novel 1H-1,2,3-triazole tethered C-5 substituted uracil-isatin conjugates and their cytotoxic evaluation. Eur J Med Chem, 2012; 58:153–9. CrossRef
Li DZ, Zhang QZ, Wang CY, Zhang YL, Li XY, Huang JT, Liu HY, Fu ZD, Song HX, Lin JP, Ji TF. Synthesis and antitumor activity of novel substituted uracil-1′(N)-acetic acid ester derivatives of 20(S)-camptothecins. Eur J Med Chem, 2017; 125:1235–46. CrossRef
Li J, Wang X, Hou J, Huang Y, Zhang Y, Xu W. Enhanced anticancer activity of 5-FU in combination with Bestatin: evidence in human tumor-derived cell lines and an H22 tumor-bearing mouse. Drug Discov Ther, 2015; 9(1):45–52. CrossRef
Liu YQ, Dai W, Yang L, Li HY. Design and synthesis of novel camptothecin/5-fluorouracil conjugates as cytotoxic agents. Nat Prod Res, 2011; 25(19):1817–26. CrossRef
Mizuno CS, Paul S, Suh N, Rimando AM. Synthesis and biological evaluation of retinoid-chalcones as inhibitors of colon cancer cell growth. Bioorg Med Chem Lett, 2010; 20(24):7385–7. CrossRef
Mo WB, Su CH, Huang JY, Liu J, Chen ZF, Cheng KG. Synthesis of acyl oleanolic acid-uracil conjugates and their anti-tumor activity. Chem Cent J, 2016; 10(1):1–11. CrossRef
Mourad MAE, Abdel-Aziz M, Abuo-Rahma GEDAA, Farag HH. Design, synthesis and anticancer activity of nitric oxide donating/chalcone hybrids. Eur J Med Chem, 2012; 54:907–13. CrossRef
Nencka R, Votruba I, HÅ™ebabecký H, Tloušt’ová E, Horská K, Masojídková M, Holý A. Design and synthesis of novel 5,6-disubstituted uracil derivatives as potent inhibitors of thymidine phosphorylase. Bioorganic Med Chem Lett, 2006; 16(5):1335–7. CrossRef
Newkome GR, Pandler WW. Contemporary heterocyclic chemistry, synthesis, reactions and applications. New York, NY: Wiley and Sons, Inc., p 785, 1982.
Nguyen HT, Lallemand MC, Boutefnouchet S, Michel S, Tillequin F. Antitumor psoropermum xanthones and sarcomelicope acridones: privileged structures implied in DNA alkylation. J Nat Prod, 2009; 72(3):527–39. CrossRef
Ota K, Kurita S, Yamada K, Masaoka T, Uzuka Y, Ogawa N. Immunotherapy with bestatin for acute nonlymphocytic leukemia in adults. Cancer Immunol Immunother, 1986; 23(1):5–10. CrossRef
Ota K, Uzuka Y. Clinical trials of bestatin for leukemia and solid tumors. Biotherapy, 1992; 4(3):205–14. CrossRef
PaÅ‚asz A, Ciez D. In search of uracil derivatives as bioactive agents. Uracils and fused uracils: Synthesis, biological activity and applications. Eur J Med Chem, 2015; 97(1):582–611. CrossRef
Pan X, Wang C, Wang F, Li P, Hu Z, Shan Y, Zhang J. Development of 5-Fluorouracil derivatives as anticancer agents. Curr Med Chem, 2011; 18(29):4538–56. CrossRef
Pettit GR, Rhodes MR, Herald DL, Hamel E, Schmidt JM, Pettit RK. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)- and (E)-combretastatin A-41. J Med Chem, 2005; 48(12):4087–99. CrossRef
Pollier J, Goossens A. Oleanolic acid. Phytochemistry, 2012; 77:10–5. CrossRef
Prakash S, Swaminathan U. β catenin in health: a review. J Oral Maxillofac Pathol, 2015; 19:230–8. CrossRef
Radwan AA, Alanazi FK. Design and synthesis of new cholesterol-conjugated 5-fluorouracil: a novel potential delivery system for cancer treatment. Molecules, 2014; 19(9):13177–87. CrossRef
Saiki I, Fujii H, Yoneda J, Abe F, Nakajima M, Tsuruo T, Azuma I. Role of aminopeptidase N (CD13) in tumor-cell invasion and extracellular matrix degradation. Int J Cancer, 1993; 54(1):137–43. CrossRef
Shen L, Hu J, Wang H, Wang A, Lai Y, Kang Y. Synthesis and biological evaluation of novel uracil and 5-fluorouracil-1-yl acetic acid-colchicine conjugate. Chem Res Chinese Univ, 2015; 31(3):367–71. CrossRef
Singh A, Mehra V, Sadeghiani N, Mozaffari S, Parang K, Kumar V. Ferrocenylchalcone–uracil conjugates: synthesis and cytotoxic evaluation. Med Chem Res [Internet], 2018; 27(4):1260–8. CrossRef
Singh P, Sharma P, Anand A, Bedi PM, Kaur T, Saxena AK, Kumar V. Azide-alkyne cycloaddition en route to novel 1H-1,2,3-triazole tethered isatin conjugates with in vitro cytotoxic evaluation. Eur J Med Chem, 2012; 55:455–61. CrossRef
Skiba J, Kowalski K, Prochnicka A, Ott I, Solecka J, Rajnisz A, Therrien B. Metallocene-uracil conjugates: Synthesis and biological evaluation of novel mono-, di- and tri-nuclear systems. J Organomet Chem, 2015; 782:52–61. CrossRef
Solano JD, González-Sánchez I, Cerbón MA, Guzmán Á, Martínez-Urbina MA, Vilchis-Reyes MA, Martínez-Zuñiga EC, Alvarado C, Quintero A, Díaz E. The products of the reaction between 6-amine-1,3-dimethyl uracil and bis-chalcones induce cytotoxicity with massive vacuolation in HeLa cervical cancer cell line. Eur J Med Chem, 2013; 60:350–9. CrossRef
Stetler-Stevenson WG. Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest, 1999; 103(9):1237–41. CrossRef
Tan YL, Pigeon P, Top S, Labbe E, Buriez O, Hillard EA, Vessières A, Amatore C, Leong WK, Jaouen G. Ferrocenyl catechols: synthesis, oxidation chemistry and anti-proliferative effects on MDA-MB-231 breast cancer cells. Dalton Trans, 2012; 41:7537–49. CrossRef
Van Hinsbergh VWM, Koolwijk P. Endothelial sprouting and angiogenesis: Matrix metalloproteinases in the lead. Cardiovasc Res, 2008; 78(2):203–12. CrossRef
Wall ME, Wani MC, Cook CE, Palmer KE, McPhail AT, Sim GA. Plant antitumor agents I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc, 1966; 88(16):3888–90. CrossRef
WHO. Latest global cancer data: Cancer burden rises to 18.1 million new cases and 9.6 million cancer deaths in 2018 [Internet]. Press Release N 263. pp 1–3, 2018. Available from http://gco.iarc.fr/ (Accessed 24 September 2019).
Xiang R, Guan XW, Hui L, Jin YX, Chen SW. Investigation of the anti-angiogenesis effects induced by deoxypodophyllotoxin-5-FU conjugate C069 against HUVE cells. Bioorganic Med Chem Lett, 2017; 27(4):713–7. CrossRef
Xiao M, Fan X, Fu Y, Zhou Y, Liu S, Peng S. Deoxypodophyllotoxin induces cell cycle arrest and apoptosis in human cholangiocarcinoma cells. Oncol Lett, 2018; 16(3):3177–82. CrossRef
Xu Z, Zhao SJ, Lv ZS, Gao F, Wang Y, Zhang F, Bai L, Deng JL. Fluoroquinolone-isatin hybrids and their biological activities. Eur J Med Chem, 2019; 396–406. CrossRef
Yen CF, Kalunta CI, Chen FS, Kaptein JS, Lin CK, Lad PM. Regulation of low-density lipoprotein receptors and assessment of their functional role in Burkitt’s lymphoma cells. Biochim Biophys Acta, 1995; 1257:47–57. CrossRef
Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules, 2008; 13(8):1551–69. CrossRef
Zhao LM, Zhang LM, Liu JJ, Wan LJ, Chen YQ, Zhang SQ, Yan ZW, Jiang JH. Synthesis and antitumor activity of conjugates of 5-Fluorouracil and emodin. Eur J Med Chem, 2012; 47(1):255–60. CrossRef
Zhu SL, Wu Y, Liu CJ, Wei CY, Tao JC, Liu HM. Design and stereoselective synthesis of novel isosteviol-fused pyrazolines and pyrazoles as potential anticancer agents. Eur J Med Chem, 2013; 65:70–82. CrossRef