
INTRODUCTION
Cancer development process basically results from the uncontrolled growth of mutated or abnormal body cells, whereas normal cells are under strict control of various growth pattern checkpoints, the neoplastic cells typically do not obey these rules and are continuously and uncontrollably proliferating. Normally, during the process of mitosis, any abnormalities during cell division are controlled by a set of physiological processes ‘apoptosis’ ending up in damaged cell death (Spano et al., 2016). Nevertheless, if the cells are mutated and not controlled by the programmed cell death and/or acquire the ability to escape several checkpoints, they turn to be malignant cells that are able to invade and metastasize (Kalyanaraman, 2017).
Various compounds have been studied as anticancer agents throughout the work on the different phases of the cell cycle. These compounds include at most the N-containing heterocycles (Akhtar et al., 2017), among which imidazole ring system represents the main core structure. Imidazole and its derivatives have a great prevalence in both natural products and synthetic molecules. Having the unique structural features with the desirable electron-rich characteristic enables these compounds to readily bind with different enzymes and receptors, thereby exhibiting broad bioactivities (Zhang et al., 2014). Various activities including anticancer, anti-inflammatory (Rapolu et al., 2013), antimicrobial (Premakumari et al., 2014), cardio-activity, and angiotensin-II receptor antagonistic activity have been depicted specifically in compounds containing imidazolone moiety (Padmaja et al., 2011). Furthermore, many imidazolones have been used as biotin antagonists capable of inhibiting the growth of malignant tumors like compound I (AK et al., 2017). For instance, the cytotoxic activity of imidazolone derivatives I and II against cervical cancer (HeLa), breast cancer (MCF-7), leukemia cells (HL-60), and hepatocellular carcinoma (HepG2) cell lines were characterized in vitro (AK et al., 2017; Kumar et al., 2017). Besides, the 2-aminoimidazolone III showed great potency toward HCT116 and H460 cell lines (IC50 of 1 and 2 μM, respectively) (Dražić et al., 2015) (Fig. 1).
Imidazolones linked to chalcone moiety were shown to have excellent antioxidant and anticancer activities on various cell lines (Mahapatra et al., 2015; Mai et al., 2014; Ramaiah et al., 2011), like compound IV (Kamal et al., 2010) that possess great potency against HCT116 and MCF-7 cell lines with GI50 values of 1.40 and 1.55 μM, respectively. It has been proved that imidazolone hybridized with chalcone moiety displayed potential antineoplastic activity through different biological mechanisms (Nasir Abbas Bukhari et al., 2013). Certain derivatives cause distinctive DNA damage specifically at telomeres and subsequently cause apoptosis (Ramaiah et al., 2011). Further mechanisms included assorted protein kinase inhibition (CDK, PLK1, MK-2, and Eg5), Topo (I, II), as well as microtubular inhibition (Akhtar et al., 2017). Moreover, they show p21 up-regulation and control damage pathway of DNA (Ramaiah et al., 2011; Yin et al., 2016).
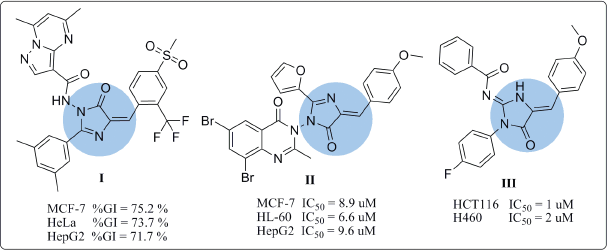 | Figure 1. Reported antitumor agents with imidazolone nucleus. [Click here to view] |
Based on the preceded data, we aim at functionalizing imidazolone-based scaffold in the design and synthesis of novel derivatives to be evaluated as anticancer agents, and exploring the plausible mechanisms by which they could exert such activity. In the current study, compound IV was chosen as the lead compound to design the new imidazolone derivatives. Rational design depended on the maintenance of the basic structural features characterizing the lead compound, mainly the imidazolone core, by replacing the 2-imidazolone ring by 4-substituted-5-imidazolone in all the designed series. Besides, optimizing the whole scaffold throughout either substitution of certain groups or replacement with various moieties, including either five or six-membered heterocyclic moieties, was accomplished to investigate the potential effect on anti-tumoral activity (Fig. 2).
MATERIALS AND METHODS
Melting points (°C) were measured with Fisher–Johns melting point apparatus and are uncorrected. Microanalyses (C, H, and N) were performed in the Microanalytical unit, Cairo University, and all the results were within ±0.5. IR spectra were recorded on Thermo Fisher Scientific Nicolet IS10 spectrometer (ν in cm‒1) using KBr disk at Faculty of Pharmacy, Mansoura University. Mass MS (EI) m/z analyses were performed on Thermo Scientific DCQII at Faculty of Science, Mansoura University. 1H and 13C-NMR spectra were achieved in dimethyl sulfoxide (DMSO)-d6 on ASCEND spectrometer (400 MHz) Bruker using TMS (chemical shifts in ppm, δ units) at Georgia State University USA, Kafrelsheikh University, and at Mansoura University. Reaction times were determined using TLC on Silica gel plates 60 F254 (E. Merk), and UV (366, 245 nm) was used to visualize the spots.
2-(4-Fluorobenzoylamino) acetic acid (1) was prepared according to the reported procedure. Yield 75% (reported 84%) and mp 168°C–170°C (reported 167°C–169°C) (El-Araby et al., 2012).
General procedure for synthesis of compounds 2 and 3
A mixture of compound 1 (1 mmol, 0.197 g), the appropriate heterocyclic aldehyde (1 mmol), and freshly-fused sodium acetate (0.049 g) in acetic anhydride (1.97 ml) was refluxed overnight at 130°C. The obtained crystals were filtered, washed with cold water, followed by aq. EtOH and recrystallized from EtOH.
2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)oxazol-5(4H)-one (2)
Yield 68%; mp 165°C–167°C; 1H NMR (DMSO d6): 6.87–7.02 (m, 3H, Ar-H), 7.44 (s, 1H, olefinic-H), and 7.61–7.93 (m, 4H, Ar-H); MS (EI) m/z %: 257 (45.5, M+); IR cm‒1: 1,775 (C=O), 1,650 (C=N); Anal. calcd for C14H8FNO3: C, 65.37; H, 3.13; and N, 5.45. Found: C, 65.29; H, 3.02; and N, 5.32.
(E)-2-(4-Fluorophenyl)-4-(thiophen-2-ylmethylene)oxazol-5(4H)-one (3)
Yield 70%; mp 173°C–175°C; 1H NMR (DMSO d6): 7.16–7.28 (m, 3H, Ar-H), 7.48 (s, 1H, olefinic-H), and 7.54–7.76 (m, 4H, Ar-H); MS (EI) m/z %: 274 (2.6, M+ + 1), 273 (4.2, M+); IR cm‒1: 1,780 (C=O), 1,656 (C=N); Anal. calcd for C14H8FNO2S: C, 61.53; H, 2.95; and N, 5.13. Found: C, 61.11; H, 2.45; and N, 5.11.
General procedure for synthesis of compounds 4–17
A mixture of compound 2 or 3 (1 mmol), the appropriate aromatic amine (1 mmol), and freshly-fused Na-acetate (0.3 g) in glacial acetic acid (7 ml) was refluxed overnight in boiling water bath, maintaining continuous stirring. The product was filtered, then washed with aq. EtOH, and recrystallized from EtOH.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-phenyl-1H-imidazol-5(4H)-one (4)
Yield 68%; mp 148°C–150°C; 1H NMR (DMSO d6): 6.85 (s, 1H, olefinic-H), 7.15–7.25 (m, 3H, Ar-H), 7.35–7.43 (m, 5H, Ar-H), and 7.75–8.15 (m, 4H, Ar-H); MS (EI) m/z %: 332 (M+), 333 (M+ + 1); IR cm‒1: 1,773 (C=O), 1,651 (C=N); Anal. calcd for C20H13FN2O2: C, 72.28; H, 3.94; and N, 8.43. Found: C, 72.19; H, 3.86; and N, 8.25.
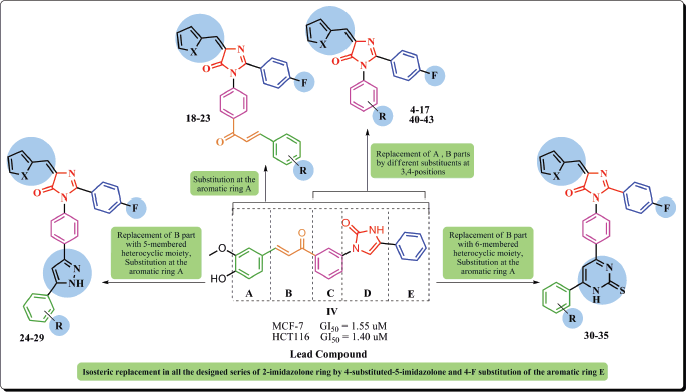 | Figure 2. Molecular design of new imidazolones as anticancer agents based on structural modification of the lead compound IV. [Click here to view] |
(E)-1-(4-Bromophenyl)-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (5)
Yield 60%; mp 135°C–137°C; 1H NMR (DMSO d6): 7.15 (s, 1H, olefinic-H), 7.35–7.78 (m, 3H, Ar-H), 7.85–8.10 (m, 4H, Ar-H), and 8.25–8.65 (m, 4H, Ar-H); MS (EI) m/z %: 410 (M+), 411 (M+ + 1); IR cm‒1: 1,768 (C=O), 1,645 (C=N); Anal. calcd for C20H12BrFN2O2: C, 5841; H, 2.94; and N, 6.81. Found: C, 58.36; H, 2.89; and N, 6.75.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-(4-methylphenyl)-1H-imidazol-5(4H)-one (6)
Yield 55% ; mp 130°C–132°C; 1H NMR (DMSO d6): 2.45 (s, 3H, CH3), 6.85 (s, 1H, olefinic-H), 7.18–7.35 (m, 3H, Ar-H), 7.45–7.64 (m, 4H, Ar-H), and 7.85–8.15 (m, 4H, Ar-H); MS (EI) m/z %: 346 (M+), 347 (M+ + 1); IR cm‒1: 1,759 (C=O), 1,647 (C=N); Anal. calcd for C21H15FN2O2: C, 72.82; H, 4.37; and N, 8.09. Found: C, 72.75; H, 4.25; and N, 8.15.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-(3-nitrophenyl)-1H-imidazol-5(4H)-one (7)
Yield 70%; mp 145°C–147°C; 1H NMR (DMSO d6): 7.15 (s, 1H, olefinic-H), 7.23–7.36 (m, 3H, Ar-H), 7.65–7.82 (m, 4H, Ar-H), and 8.43–8.77 (m, 4H, Ar-H); MS (EI) m/z %: 377 (M+), 378 (M+ + 1); IR cm‒1: 1,757 (C=O), 1,644 (C=N); Anal. calcd for C20H12FN3O4: C, 63.66; H, 3.21; and N, 11.14. Found: C, 63.54; H, 3.15; and N, 11.20.
(E)-1-(3-Acetylphenyl)-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (8)
Yield 65%; mp 125°C–127°C; 1H NMR (DMSO d6): 2.35 (s, 3H, CH3), 7.13 (s, 1H, olefinic-H), 7.18–7.37 (m, 3H, Ar-H), 7.45–7.87 (m, 4H, Ar-H), and 7.95–8.15 (m, 4H, Ar-H); MS (EI) m/z %: 374 (M+), 375 (M+ + 1); IR cm‒1: 1,750 (C=O), 1,652 (C=N); Anal. calcd for C22H15FN2O3: C, 70.58; H, 4.04; and N, 7.37. Found: C, 70.45; H, 4.02; and N, 7.37.
(E)-1-(4-Acetylphenyl)-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (9)
Yield 70%; mp 130°C–132°C; 1H NMR (DMSO d6): 2.54 (s, 3H, CH3), 7.12 (s, 1H, olefinic-H), 7.23–7.36 (m, 4H, Ar-H), and 7.76–7.89 (m, 4H, Ar H); MS (EI) m/z %: 374 (M+), 375 (M+ + 1); IR cm‒1: 1,761 (C=O) imidazolone, 1,685 (C=O) acetyl, 1,658 (C=N); Anal. calcd for C22H15FN2O3 : C, 70.58; H, 4.04; and N, 7.37. Found: C, 70.48; H, 4.02; and N, 7.32.
(E)-Ethyl 4-[2-(4-fluorophenyl)]-4-(furan-2-ylmethylene)-5-oxo-4,5-dihydro-1H-imidazol-1-yl)benzoate (10)
Yield 65%; mp 170°C–173°C; 1H NMR (DMSO d6): 1.50 (t, 3H, CH3), 4.35 (q, 2H, CH2), 7.15 (s, 1H, olefinic-H), 7.20–7.35 (m, 3H, Ar-H), 7.44–7.67 (m, 4H, Ar-H), and 7.78–7.98 (m, 4H, Ar-H); MS (EI) m/z %: 404 (M+), 405(M+ + 1); IR cm‒1: 1,765 (C=O) imidazolone, 1,727 (C=O) ester, 1,657 (C=N); Anal. calcd for C23H17FN2O4: C, 68.31; H, 4.24; and N, 6.93. Found: C, 68.23; H, 4.18; and N, 6.85.
(E)-2-(4-Fluorophenyl)-1-phenyl-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (11)
Yield 71%; mp 100°C–102°C; 1H NMR (DMSO d6): 7.14 (s, 1H, olefinic H), 7.23–7.34 (m, 3H, Ar-H), 7.45–7.58 (m, 5H, Ar-H), and 7.75–8.15 (m, 4H, Ar-H); MS (EI) m/z %: 348 (M+), 349 (M+ + 1); IR cm‒1: 1,767 (C=O), 1,662 (C=N); Anal. calcd for C20H13FN2OS: C, 68.95; H, 3.76; and N, 8.04. Found: C, 68.85; H, 3.65; and N, 8.02.
(E)-1-(4-Bromophenyl)-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (12)
Yield 65%; mp 140°C–142°C; 1H NMR (DMSO d6): 7.19 (s, 1H, olefinic H), 7.43–7.78 (m, 3H, Ar-H), 7.87–8.10 (m, 4H, Ar-H), and 8.36–8.65 (m, 4H, Ar-H); MS (EI) m/z %: 425 (M+), 426 (M+ + 1); IR cm‒1: 1,762 (C=O), 1,671 (C=N); Anal. calcd for C20H12BrFN2OS: C, 56.22; H, 2.83; and N, 6.56. Found: C, 56.15; H, 2.74; and N, 6.45.
(E)-2-(4-Fluorophenyl)-4-(thiophen-2-ylmethylene)-1-(4-methylphenyl)-1H-imidazol-5(4H)-one (13)
Yield 75% ; mp 163°C–167°C; 1H NMR (DMSO d6): 2.35 (s, 3H, CH3), 6.88 (s, 1H, olefinic), 7.23–7.35 (m, 3H, Ar-H), 7.56–7.64 (m, 4H, Ar-H), and 7.85–8.12 (m, 4H, Ar-H); MS (EI) m/z %: 362 (M+), 363 (M+ + 1); IR cm‒1: 1,762 (C=O), 1,669 (C=N); Anal. calcd for C21H15FN2OS: C, 69.59; H, 4.17; and N, 7.73. Found: C, 69.4; H, 4.20; and N, 7.67.
(E)-2-(4-Fluorophenyl)-1-(3-nitrophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (14)
Yield 68%; mp 142°C–145°C; 1H NMR (DMSO d6): 7.15 (s, 1H, olefinic H), 7.23–7.45 (m, 3H, Ar-H), 7.65–7.82 (m, 4H, Ar-H), and 8.36–8.77 (m, 4H, Ar-H); MS (EI) m/z %: 393 (M+), 394 (M+ + 1); IR cm‒1: 1,765 (C=O), 1,666 (C=N); Anal. calcd for C20H12FN3O3S: C, 61.06; H, 3.07; and N, 10.68. Found: C, 61.15; H, 3.11; and N, 10.54.
(E)-1-(3-Acetylphenyl)-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (15)
Yield 71%; mp 120°C–122°C; 1H NMR (DMSO d6): 2.53 (s, 3H, CH3), 6.98 (s, 1H, olefinic-H), 7.02–7.14 (m, 3H, Ar H), 7.34–7.56 (m, 4H, Ar H), and 7.75–7.91 (m, 4H, Ar H); MS (EI) m/z %: 390 (M+), 391 (M+ + 1); IR cm‒1: 1,768 (C=O), 1,684 (C=O) acetyl, 1,657 (C=N); Anal. calcd for C22H15FN2O2S: C, 67.68; H, 3.87; and N, 7.18. Found: C, 67.54; H, 3.75; and N, 7.11.
(E)-1-(4-Acetylphenyl)-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (16)
Yield 75%; mp 132°C–134°C; 1H NMR (DMSO d6): 2.52 (s, 3H, CH3), 6.92 (s, 1H, olefinic-H), 7.23–7.41 (m, 8H, Ar-H), and 7.93–8.11 (m, 3H, Ar-H); 13C NMR (DMSO d6): Δ 20.7, 89.4, 100.6, 110.5, 117.0, 120.3, 122.5, 127.5, 130.5, 132.3, 137.3, 140.8, 144.6, 148.3, 153.9, 156.2, 158.0, 166.0, 169.6, 177.6, and 190.1; MS (EI) m/z %: 390 (M+), 391 (M+ + 1); IR cm‒1: 1,768 (C=O) imidazolone, 1,687 (C=O) acetyl, 1,655 (C=N); Anal. calcd for C22H15FN2O2S: C, 67.68; H, 3.87; and N, 7.18. Found: C, 67.58; H, 3.45; and N, 7.09.
(E)-Ethyl 4-[2-(4-fluorophenyl)-5-oxo-4-(thiophen-2-ylmethylene)-4,5-dihydro-1H-imidazol-1-yl]benzoate (17)
Yield 69%; mp 167°C–170°C; 1H NMR (DMSO d6): 1.21 (t, 3H, CH3), 4.27 (q, 2H, CH2), 6.96 (s, 1H, olefinic-H), 7.08–7.28 (m, 4H, Ar H), and 7.43–7.98 (m, 7H, Ar H); 13CNMR (DMSO d6): Δ 10.7, 25.4, 100.2, 113.5, 114.3, 117.4, 118.5, 120.6, 123.7, 124.2, 125.7, 126.5, 127.4, 130.3, 133.5, 142.3, 147.4, 152.7, 153.9, 155.3, 160.4, 166.7, and 200.0, MS (EI) m/z %: 420 (17.5, M+); IR cm‒1: 1,759 (C=O) imidazolone, 1,725 (C=O) ester, 1,649 (C=N); Anal. calcd for C23H17FN2O3S: C, 65.70; H, 4.08; and N, 6.66. Found: C, 65.45; H, 4.02; and N, 6.55.
General procedure for synthesis of compounds 18–23
A mixture of compound 9 or 19 (1 mmol) and different aromatic aldehydes (1 mmol) was stirred in ethanolic sodium hydroxide solution (5%, 10 ml) in ice bath for 4 hours, then the resulted precipitate was filtered, washed with aq. EtOH, and recrystallized from EtOH.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-[4-(3-phenylprop-2-enoyl)phenyl]-1H-imidazol-5(4H)-one (18)
Yield 50%; mp 118°C–120°C; 1H NMR (DMSO d6): 6.89 (s, 1H, oleifinic H), 7.18–7.23 (m, 3H, Ar-H), 7.50 (d, 1H, CH=CH), 7.78 (d, 1H, CH=CH), 8.12–8.23 (m, 4H, Ar-H), 8.35–8.47 (m, 4H, Ar-H), and 8.76–9.11 (m, 5H, Ar-H); MS (EI) m/z %: 462.14 (M+), 463 (M+ + 1); IR cm‒1: 1,765 (C=O), 1,655 (C=N); Anal. calcd for C29H19FN2O3: C, 75.32; H, 4.14; and N, 6.06. Found: C, 75.32; H, 4.25; and N, 6.15.
(E)-{4-[3-(3-Bromophenyl)prop-2-enoyl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (19)
Yield 65%; mp 115°C–117°C; 1H NMR (DMSO d6): 7.14 (s, 1H, oleifinic H), 7.32–7.45 (m, 3H, Ar-H), 7.64 (d, 1H, CH=CH), 7.85 (d, 1H, CH=CH), 8.32–8.45 (m, 4H, Ar-H), 8.67–8.98 (m, 4H, Ar-H), and 9.00–9.10 (m, 4H, Ar-H); MS (EI) m/z %: 540 (M+), 541 (M+ + 1); IR cm‒1: 1,750 (C=O), 1,661 (C=N); Anal. calcd for C29H18BrFN2O3: C, 64.34; H, 3.35; and N, 5.17. Found: C, 64.25; H, 3.15; and N, 5.26.
(E)-{4-[3-(4-Chlorophenyl)prop-2-enoyl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (20)
Yield 69%; mp 120°C–122°C; 1H NMR (DMSO d6): 7.04 (s, 1H, oleifinic-H), 7.27–7.36 (m, 3H, Ar-H), 7.78 (d, 1H, CH=CH), 7.98 (d, 1H, CH=CH), 8.15–8.34 (m, 4H, Ar-H), 8.48–8.67 (m, 4H, Ar-H), and 8.78–9.89 (m, 4H, Ar-H); MS (EI) m/z %: 496.10 (M+), 497 (M+ + 1); IR cm‒1: 1,755 (C=O), 1,660 (C=N); Anal. calcd for C29H18ClFN2O3: C, 70.09; H, 3.65; and N, 5.64. Found: C, 70.45; H, 3.45; and N, 5.87.
(E)-2-(4-Fluorophenyl)-4-(thiophen-2-ylmethylene)-1-[4-(3-phenylprop-2-enoyl)phenyl]-1H-imidazol-5(4H)-one (21)
Yield 55%; mp 125°C–127°C; 1H NMR (DMSO d6): 7.36 (s, 1H, oleifinic-H), 7.43 (d, 1H, CH=CH), 8.16 (d, 1H, CH=CH), 8.32–8.45 (m, 4H, Ar-H), 8.67–8.98 (m, 4H, Ar-H), and 9.01–9.10 (m, 5H, Ar-H); MS (EI) m/z %: 478.54 (M+); IR cm‒1: 1,757 (C=O), 1,656 (C=N); Anal. calcd for C29H19FN2O2S: C, 72.79; H, 4; and N, 5.85. Found: C, 72.45; H, 4.15; and N, 5.45.
(E)-{4-[3-(3-Bromophenyl)prop-2-enoyl]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (22)
Yield 60%; mp 114°C–116°C; 1H NMR (DMSO d6): 7.36 (s, 1H, oleifinic-H), 7.59 (d, 1H, CH=CH), 8.23 (d, 1H, CH=CH), 8.33–8.46 (m, 4H, Ar-H), 8.68–8.88 (m, 4H, Ar-H), and 9.00–9.12 (m, 4H, Ar-H); MS (EI) m/z %: 557.43 (M+); IR cm‒1: 1,761 (C=O), 1,650 (C=N); Anal. calcd for C29H18BrFN2O2S: C, 62.48; H, 3.25; and N, 5.03. Found: C, 62.35; H, 3.15; and N, 5.15.
(E)-{4-[3-(4-Chlorophenyl)prop-2-enoyl]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (23)
Yield 67%; mp 124°C–126°C; 1H NMR (DMSO d6): 7.09 (s, 1H, olefinic-H), 7.87 (d, 1H, CH=CH), 8.43 (d, 1H, CH=CH), 8.32–8.45 (m, 4H, Ar-H), 8.67–8.98 (m, 4H, Ar-H), and 9.02–9.10 (m, 4H, Ar-H); MS (EI) m/z %: 512.98 (M+); IR cm‒1: 1,755 (C=O), 1,657 (C=N); Anal. calcd for C29H18ClFN2O2S: C, 67.9; H, 3.54; and N, 5.46. Found: C, 67.85; H, 3.34; and N, 5.67.
General procedure for synthesis of compounds 24–29
A mixture of the appropriate chalcone 18–23 (1mmol) and hydrazine hydrate 98% (1 mmol, 0.03 ml) in abs. EtOH (5 ml) was refluxed overnight followed by evaporation of the solvent, and then the product was neutralized with dil. HCl. The precipitated product was filtered and dried after washing thoroughly with water.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-[4-(5-phenyl-1H-pyrazol-3-yl)phenyl]-1H-imidazol-5(4H)-one (24)
Yield 50%; mp 217°C–220°C; 1H NMR (DMSO d6): 6.51 (s, 1H, pyrazole-H), 6.89 (s, 1H, olefinic-H), 7.12–7.32 (m, 3H, Ar-H), 7.84–8.11 (m, 4H, Ar-H), 8.38–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 5H, Ar-H), and 12.45 (s, 1H, NH); 13CNMR (DMSO d6): Δ 89.5, 110.4, 112.2, 114.5, 115.4, 117.7, 119.5, 120.5, 121.7, 122.9, 123.4, 125.4, 126.6, 127.9, 128.5, 129.8, 130.0, 132.4, 135.0, 136.4, 138.4, 139.6, 144.7, 146.3, 148.5, 152.0, 153.9, 166.4, and 170.5; MS (EI) m/z %: 474 (M+), 475 (M+ + 1); IR cm‒1: 3,340 (NH), 1,750 (C=O), 1,661 (C=N); Anal. calcd for C29H19FN4O2: C, 73.41; H, 4.04; and N, 11.81. Found: C, 73.35; H, 4.15; and N, 11.72.
(E)-1-{4-[5-(3-Bromophenyl)-1H-pyrazol-3-yl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (25)
Yield 55%; mp 198°C–200°C; 1H NMR (DMSO d6): 6.87 (s, 1H, pyrazole-H), 7.14 (s, 1H, olefinic-H), 7.24–7.65 (m, 3H, Ar-H), 7.95–8.23 (m, 4H, Ar-H), 8.48–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 4H, Ar-H), 12.45 (s, 1H, NH), and 12.64 (s, 1H, NH); MS (EI) m/z %: 552 (M+), 553 (M++1); IR cm‒1: 3,410 (NH), 1,759 (C=O), 1,655 (C=N); Anal. calcd for C29H18BrFN4O2: C, 62.94; H, 3.28; and N, 10.12. Found: C, 62.87; H, 3.15; and N, 10.15.
(E)-1-{4-[5-(4-Chlorophenyl)-1H-pyrazol-3-yl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (26)
Yield 56%; mp 158°C–160°C; 1H NMR (DMSO d6): 7.04 (s, 1H, pyrazole-H), 7.23 (s, 1H, olefinic-H), 7.35–7.75 (m, 3H, Ar-H), 7.98–8.34 (m, 4H, Ar-H), 8.48–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 4H, Ar-H), 12.45 (s, 1H, NH), 12.64 (s, 1H, NH), and 12.77 (s, 1H, NH); 13CNMR (DMSO d6): Δ 87.5, 111.4, 112.3, 113.2, 115.9, 116.9, 118.5, 119.5, 120.8, 121.9, 124.7, 125.9, 126.5, 127.9, 128.3, 129.5, 130.4, 132.4, 135.0, 137.4, 138.4, 139.6, 143.7, 145.3, 147.5, 152.0, 153.9, 166.4, and 170.5; MS (EI) m/z %: 508 (M+), 509 (M++1); IR cm‒1: 3,310 (NH), 1,755 (C=O), 1,661 (C=N); Anal. calcd for C29H18ClFN4O2: C, 68.44; H, 3.56; and N, 11.01. Found: C, 68.65; H, 3.45; and N, 11.15.
(E)-2-(4-Fluorophenyl)-1-[4-(5-phenyl-1H-pyrazol-3-yl)phenyl]-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (27)
Yield 56%; mp 200°C–202°C; 1H NMR (DMSO d6): 6.45 (s, 1H, pyrazole-H), 7.12 (s, 1H, olefinic-H), 7.23–7.56 (m, 3H, Ar-H), 7.84–8.11 (m, 4H, Ar-H), 8.38–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 5H, Ar-H), 12.45 (s, 1H, NH), and 12.45 (s, 1H, NH); MS (EI) m/z %: 490 (M+), 491 (M+ + 1); IR cm‒1: 3,410 (NH), 1,754 (C=O), 1,656 (C=N); Anal. calcd for C29H19FN4OS: C, 71; H, 3.9; and N, 11.42. Found: C, 71.15; H, 3.75; and N, 11.32.
(E)-1-{4-[5-(3-Bromophenyl)-1H-pyrazol-3-yl]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (28)
Yield 59%; mp 208°C–210°C; 1H NMR (DMSO d6): 6.65 (s, 1H, pyrazole-H), 6.87 (s, 1H, olefinic-H), 7.24–7.65 (m, 3H, Ar-H), 7.95–8.23 (m, 4H, Ar-H), 8.48–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 4H, Ar-H), 12.45 (s, 1H, NH), 12.64 (s, 1H, NH), and 12.64 (s, 1H, NH); MS (EI) m/z %: 569.45 (M+); IR cm‒1: 3,330 (NH), 1,758 (C=O), 1,655 (C=N); Anal. calcd for C29H18BrFN4OS: C, 61.17; H, 3.19; and N, 9.84. Found: C, 61.23; H, 3.15; and N, 9.78.
(E)-1-{4-[5-(4-Chlorophenyl)-1H-pyrazol-3-yl]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (29)
Yield 58%; mp 160°C–162°C; 1H NMR (DMSO d6): 7.04 (s, 1H, pyrazole-H), 7.23 (s, 1H, olefinic-H), 7.35–7.75 (m, 3H, Ar-H), 7.95–8.23 (m, 4H, Ar-H), 8.48–8.76 (m, 4H, Ar-H), 8.89–9.10 (m, 4H, Ar-H), 12.45 (s, 1H, NH), 12.64 (s, 1H, NH), and 12.77 (s, 1H, NH); MS (EI) m/z %: 524 (M+), 525 (M+ + 1); IR cm‒1: 3,350 (NH), 1,757 (C=O), 1,662 (C=N); Anal. calcd for C29H18ClFN4OS: C, 66.35; H, 3.46; and N, 10.67. Found: C, 66.15; H, 3.24; and N, 10.87.
General procedure for synthesis of compounds 30–35
To the solution of thiourea (1 mmol, 0.076 g) and sodium ethoxide in ethanol (5 ml), the appropriate chalcone18–23 (1 mmol) was added. The mixture was refluxed overnight. After cooling and evaporation of the solvent, the mixture was neutralized with dil. HCl and the precipitate was filtered and washed thoroughly with water.
(E)-2-(4-Fluorophenyl)-4-(furan-2-ylmethylene)-1-[4-(6-phenyl-2-thioxo-1,2-dihydropyrimidin-4-yl)phenyl)]-1H-imidazol-5(4H)-one (30)
Yield 53%; mp 215°C–217°C; 1H NMR (DMSO d6): 6.23 (s, 1H, pyrazole-H), 7.13 (s, 1H, olefinic-H), 7.34–7.65 (m, 3H, Ar-H), 7.75–7.89 (m, 4H, Ar-H), 8.15–8.23 (m, 4H, Ar-H), 8.38–8.45 (m, 5H, Ar-H), 12.45 (s, 1H, NH), 12.64 (s, 1H, NH), and 13.65 (s, 1H, NH); MS (EI) m/z %: 518 (M+), 519 (M+ + 1); IR cm‒1: 3,310 (NH), 1,760 (C=O), 1,654 (C=N); Anal. calcd for C30H19FN4O2S: C, 69.48; H, 3.69; and N, 10.08. Found: C, 69.54; H, 3.59; and N, 10.76.
(E)-1-{4-[6-(3-Bromophenyl)-2-sulfanylidene-1,2-dihydropyrimidin-4-yl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (31)
Yield 56%; mp 218°C–220°C; 1H NMR (DMSO d6): 6.65 (s, 1H, pyrazole-H), 7.25 (s, 1H, olefinic-H), 7.34–7.78 (m, 3H, Ar-H), 7.89–8.14 (m, 4H, Ar-H), 8.33–8.68 (m, 4H, Ar-H), 8.79–8.89 (m, 4H, Ar-H), and 13.45 (s, 1H, NH); MS (EI) m/z %: 596 (M+), 597 (M+ + 1); IR cm‒1: 3,310 (NH), 1,758 (C=O), 1,652 (C=N); Anal. calcd for C30H18BrFN4O2S: C, 60.31; H, 3.04; and N, 9.38. Found: C, 60.25; H, 3.54; and N, 9.48.
(E)-1-{4-[6-(4-Chlorophenyl)-2-sulfanylidene-1,2-dihydropyrimidin-4-yl]phenyl}-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (32)
Yield 56%; mp 162°C–164°C; 1H NMR (DMSO d6): 7.02 (s, 1H, pyrazole-H), 7.35 (s, 1H, olefinic-H), 7.44–7.78 (m, 3H, Ar-H), 7.98–8.17 (m, 4H, Ar-H), 8.37–8.58 (m, 4H, Ar-H), 8.79–8.89 (m, 4H, Ar-H), and 13.45 (s, 1H, NH); MS (EI) m/z %: 552 (M+), 553 (M+ + 1); IR cm‒1: 3,310 (NH), 1,762 (C=O), 1,659 (C=N); Anal. calcd for C30H18ClFN4O2S: C, 65.16; H, 3.28; and N, 10.13. Found: C, 65.25; H, 3.15; and N, 10.27.
(E)-2-(4-Fluorophenyl)-1-[4-(6-phenyl-2-sulfanylidene-1,2-dihydropyrimidin-4-yl)phenyl]-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (33)
Yield 55%; mp 200°C–203°C; 1H NMR (DMSO d6): 6.23 (s, 1H, pyrazole-H), 7.13 (s, 1H, olefinic-H), 7.34–7.65 (m, 3H, Ar-H), 7.75–7.89 (m, 4H, Ar-H), 8.15–8.23 (m, 4H, Ar-H), 8.38–8.45 (m, 5H, Ar-H), and 13.65 (s, 1H, NH); MS (EI) m/z %: 534 (M+), 544 (M+ + 1); IR cm‒1: 3,320 (NH), 1,757 (C=O), 1,665 (C=N); Anal. calcd for C30H19FN4OS2: C, 67.4; H, 3.58; and N, 10.48. Found: C, 67.65; H, 3.45; and N, 10.35.
(E)-1-{4-[6-(3-Bromophenyl)-2-sulfanylidene-1,2-dihydropyrimidin-4-yl]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (34)
Yield 59%; mp 215°C–217°C; 1H NMR (DMSO d6): 6.65 (s, 1H, pyrazole-H), 7.25 (s, 1H, olefinic-H), 7.34–7.78 (m, 3H, Ar-H), 7.89–8.14 (m, 4H, Ar-H), 8.33–8.68 (m, 4H, Ar-H), 8.79–8.89 (m, 4H, Ar-H), and 13.45 (s, 1H, NH); MS (EI) m/z %: 612 (M+), 613 (M+ + 1); IR cm‒1: 3,310 (NH), 1,753 (C=O), 1,651 (C=N); Anal. calcd for C30H18BrFN4OS: C, 58.73; H, 2.96; and N, 9.13. Found: C, 58.65; H, 2.45; and N, 9.15.
(E)-1-{4-[6-(4-Chlorophenyl)-2-sulfanylidene-1,2-dihydropyrimidin-4-yl)]phenyl}-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (35)
Yield 58%; mp 168°C–170°C; 1H NMR (DMSO d6): 7.02 (s, 1H, pyrazole-H), 7.35 (s, 1H, olefinic-H), 7.44–7.78 (m, 3H, Ar-H), 7.98–8.17 (m, 4H, Ar-H), 8.37–8.58 (m, 4H, Ar-H), 8.79–8.89 (m, 4H, Ar-H), and 13.45 (s, 1H, NH); MS (EI) m/z %: 568 (M+), 569 (M+ + 1); IR cm‒1: 3,310 (NH), 1,760 (C=O), 1,662 (C=N); Anal. calcd for C30H18ClFN4OS: C, 67.10; H, 3.38; and N, 10.43. Found: C, 67.23; H, 3.28; and N, 10.41.
General procedure for synthesis of compounds 36–39
A solution of the appropriate acetyl derivative (8, 9, 15, 16) (1 mmol) in glacial acetic acid (15 ml) was heated to 80°C–90°C with stirring. Bromine (1 mmol, 0.159 g) was added drop by drop over 30 minutes to the hot mixture with stirring at 80°C–90°C. After completion of bromine addition, the mixture continued to stir at room temperature for further 1 hour till evolution of hydrogen bromide gas was ceased. Then, it was poured over ice where the formed product was filtered, washed thoroughly with water, and recrystallized from 70% EtOH.
(E)-1-[3-(2-Bromoacetyl)phenyl]-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (36)
Yield 69%; mp 133°C–137°C; 1H NMR (DMSO d6): 4.54 (s, 2H, CH2), 7.27 (s, 1H, olefinic-H), 7.56–7.90 (m, 3H, Ar-H), and 8.00–8.11 (m, 8H, Ar H); MS (EI) m/z %: 452 (M+), 453 (M+ + 1); IR cm‒1: 1,765 (C=O), 1,652 (C=N); Anal. calcd for C22H14BrFN2O3: C, 56.3; H, 3.01; and N, 5.97. Found: C, 65.25; H, 3.53; and N, 5.65.
(E)-1-[4-(2-Bromoacetyl)phenyl]-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (37)
Yield 68%; mp 125°C–128°C; 1H NMR (DMSO d6): 4.67 (s, 2H, CH2), 7.27 (s, 1H, olefinic-H), 7.56–7.78 (m, 3H, Ar-H), and 7.92–8.11 (m, 8H, Ar H); MS (EI) m/z %: 452 (M+), 453 (M+ + 1); IR cm‒1: 1,761 (C=O), 1,654 (C=N); Anal. calcd for C22H14BrFN2O3: C, 58.3; H, 3.11; and N, 6.18. Found: C, 58.25; H, 3.15; and N, 6.17.
(E)-1-[3-(2-Bromoacetyl)phenyl]-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (38)
Yield 73%; mp 130°C–133°C; 1H NMR (DMSO d6): 4.5 (s, 2H, CH2), 7.45 (s, 1H, olefinic H), 7.56–7.67 (m, 3H, Ar-H), and 7.88–8.23 (m, 8H, Ar H); 13CNMR (DMSO d6): Δ 29.4, 107.6, 114.7, 115.9, 118.7, 124.9, 125.9, 127.5, 129.5, 131.5, 132.9, 133.0, 134.7, 135.9, 136.5, 138.5, 139.3, 140.5, 158.4, 166.4, 168.2, and 200.5; MS (EI) m/z %: 467 (M+), 468 (M+ + 1); IR cm‒1: 1,750 (C=O), 1,656 (C=N); Anal. calcd for C22H14BrFN2O2S: C, 56.3; H, 3.01; and N, 5.97. Found: C, 65.25; H, 3.53; and N, 5.65.
(E)-1-[4-(2-Bromoacetyl)phenyl]-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (39)
Yield 70; mp 115°C–120°C; 1H NMR (DMSO d6): 4.3 (s, 2H, CH2), 7.45 (s, 1H, olefinic-H), 7.52–7.77 (m, 3H, Ar-H), and 7.89–8.19 (m, 8H, Ar H); MS (EI) m/z %: 467 (M+), 468 (M+ + 1); IR cm‒1: 1,762 (C=O), 1,657 (C=N); Anal. calcd for C22H14BrFN2O2S: C, 56.3; H, 3.01; and N, 5.97. Found: C, 65.25; H, 3.53; and N, 5.65.
General procedure for synthesis of compounds 40–43
To a solution of the appropriate bromo derivative (36–39) (1 mmol) in abs. EtOH (20 ml), thiourea was added (1 mmol, 0.076 g). The mixture was refluxed overnight, then it was cooled, followed by the addition of ammonium hydroxide. The precipitate was filtered and washed several times with water.
(E)-1-[3-(2-Aminothiazol-4-yl)phenyl]-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (40)
Yield 60%; mp 260°C–262°C; 1H NMR (DMSO d6): 7.11 (s, 2H, NH2), 7.77–8.11 (m, 8H, Ar H), and 7.36 (s, 1H, olefinic-H); MS (EI) m/z %:430 (% M+), 431 (M+ + 1); IR cm‒1: 3,470 (NH2), 1,753 (C=O), 1,655 (C=N); Anal. calcd for C23H15FN4O2S: C, 61.87; H, 3.39; and N, 12.55. Found: C, 61.74; H, 3.14; and N, 12.45.
(E)-1-[4-(2-Aminothiazol-4-yl)phenyl]-2-(4-fluorophenyl)-4-(furan-2-ylmethylene)-1H-imidazol-5(4H)-one (41)
Yield 69%; mp 243°C–245°C; 1H NMR (DMSO d6): 7.24 (s, 2H, NH2), 7.74–8.11 (m, 8H, Ar H), and 7.36 (s, 1H, olefinic-H); MS (EI) m/z %: 430 (% M+), 431 (M+ + 1); IR cm‒1: 3,468 (NH2), 1,761 (C=O), and 1,659 (C=N); Anal. calcd for C23H15FN4O2S: C, 64.87; H, 3.51; and N, 13.02. Found: C, 64.25; H, 3.45; and N, 13.15.
(E)-1-[3-(2-Aminothiazol-4-yl)phenyl]-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (42)
Yield 55%; mp 255°C–257°C; 1H NMR (DMSO d6): 7.17 (s, 2H, NH2), 7.89–8.11 (m, 8H, Ar H), and 7.36 (s, 1H, olefinic-H); MS (EI) m/z %: 446 (% M+), 447(M+ + 1); IR cm‒1: 3,482 (NH2), 1,763 (C=O), 1,655 (C=N); Anal. calcd for C23H15FN4OS2: C, 61.87; H, 3.39; and N, 12.55. Found: C, 61.74; H, 3.14; and N, 12.45.
(E)-1-[4-(2-Aminothiazol-4-yl)phenyl]-2-(4-fluorophenyl)-4-(thiophen-2-ylmethylene)-1H-imidazol-5(4H)-one (43)
Yield 73%; mp 256°C–258°C; 1H NMR (DMSO d6): 7.65 (s, 2H, NH2), 7.75–8.11 (m, 8H, Ar H), and 7.36 (s, 1H, olefinic-H); MS (EI) m/z %: 446 (% M+), 447 (M+ + 1); IR cm‒1: 3,475 (NH2), 1,761 (C=O), and 1,656 (C=N); Anal. calcd for C23H15FN4OS2: C, 61.87; H, 3.39; and N, 12.55. Found: C, 61.74; H, 3.14; and N, 12.45.
Biological evaluation
Preliminary in vitro cytotoxic screening
Cell lines: Four human tumor cell lines, namely, Epithelioid carcinoma (Hela), breast cancer (MCF-7), prostate cancer (PC3), and colorectal carcinoma (HCT-116) were obtained from ATCC through Holding company for biological products and vaccines (VACSERA), Cairo, Egypt.
Chemicals and reagents: The reagents used were RPMI-1640 medium, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and DMSO (Sigma co., St. Louis, USA), and Fetal Bovine Serum (GIBCO, UK). Doxorubicin was used as a standard anticancer drug for comparison.
MTT assay
Cytotoxicity of the target compounds was determined via MTT assay on different cell lines. The assay depends on the conversion of yellow tetrazolium bromide (MTT) to a purple formazan derivative by mitochondrial succinate dehydrogenase. The process is initiated by culturing the cells in fetal bovine serum (10%) mixed with RPMI-1640 medium. Both penicillin (100 units/ml) and streptomycin (100 μg/ml) were added in a 5% CO2 incubator, at a temperature of 37°C. Cells were grown at a density of 1.0 × 104 cells/well at 37°C under 5% CO2 in a 96-well plate for 48 hours. Cells were treated with various concentrations of the tested compounds after incubation, where they were further incubated for 24 hours. Thereafter, 20 μl of MTT at 5 mg/ml was added and incubated for 4 hours. The dissolution of the purple formazan formed was achieved by the addition of 100 μl of DMSO into each well. The colorimetric assay was performed and recorded at 570 nm via a microplate reader (EXL 800, USA). % Cell viability was then calculated as previously reported (Denizot and Lang, 1986; Mosmann, 1983).
CDK2A enzyme assay
Seven wells for standard and one well for blank were prepared. Add 100 μl each of dilutions of standard (read reagent preparation), blank, and samples into the appropriate wells. Cover with the plate sealer. Incubate for 2 hours at 37°C. Then, the liquid of each well was removed. After that, 100 μl of detection reagent was added to each well and incubated for 1 hour at 37°C after covering with the plate sealer, followed by washing of each well with 350 μl of the washing solution and left for 1–2 minutes. The remaining liquid was completely removed from all wells by snapping the plate onto absorbent paper, totally washed for three times, after which, any remaining wash buffer was removed, followed by plate inversion and blotting against absorbent paper. The previous process was repeated again using 100 μl of detection reagent B working solution. Ninety miroliter of substrate solution was added to each well, covered with a new plate sealer and incubated for 15–25 minutes at 37°C. The liquid was turned blue by the addition of substrate solution. Then, the microplate reader was run and measurement was conducted at 450 nm immediately. Results were calculated by determination of the duplicate reading average for each standard, control, and samples, where the average zero standard optical density was subtracted. Then, construction of the standard curve was performed by plotting the mean O.D. and concentration for each standard, and a best-fit curve was drawn through the points on the graph or standard curve on log-log graph paper with CDK2 concentration on the Y-axis and absorbance on the X-axis was created.
VEGFR-2 enzyme assay
All reagents and samples were used at room temperature (18°C–25°C). Both the standards and samples were run in duplicate, then 100 μl of each standard and sample was added to the appropriate wells and incubated overnight at 4°C with gentle shaking. The solution was washed four times with the washing solution, where any remaining wash buffer was removed. Plate inversion was performed, and then it was blotted. Each well was subjected to the addition of the biotinylated antibody (100 μl) and then incubated for 1 hour at room temperature. After washing, the addition of the streptavidin solution (100 μl) was made, followed by incubation for 45 minutes at room temperature. Also, 100 μl of TMB (Item H) was added and then incubated for 30 minutes in the dark. Finally, 50 μl of stop solution was added and read at 450 nm. The mean absorbance for each set of duplicate standards, controls, and samples was calculated, and the average zero standard optical density was subtracted. Standard curve was drawn using Sigma plot software and then the best-fit straight line was drawn through the standard points.
RESULTS AND DISCUSSION
Chemistry
Imidazolone ring has a particular synthetic interest rather than the unique biological value owing to synthetic accessibility and structural variability. Besides, it constitutes the core structure of several natural compounds like histamine, histidine, biotin, alkaloids, and nucleic acids. It has been reported to be prepared by several methods, including the condensation of substituted azalactone with primary amines under anhydrous conditions (Kortiwala et al., 2016). In the present work, the synthetic pathways adopted for the preparation of the target new imidazolone derivatives are illustrated in Schemes 1–3.
4-Fluorobenzoyl chloride was subjected to nucleophilic condensation reaction with glycine in aq. NaOH to afford hippuric acid derivative 1 as the starting material. Oxazolone derivatives 2 and 3 were synthesized via Erlenmeyer condensation (El-Mekabaty, 2013; Erlenmeyer, 1900) of compound 1 with different heterocyclic aldehydes in acetic anhydride and glacial acetic acid. Disappearance of the broad band signal at 2,970 cm-1; that corresponds to the (OH) of compound 1; in the IR spectrum of both compounds 2 and 3, in addition to the replacement of the (C=O) band at 1,720 cm-1 with 1,775-1,780 cm-1 confirmed the formation of the oxazolone ring. Subsequent reaction with different aromatic amines yielded the required imidazolone derivatives 4–17 (Scheme 1).
Chalcones 18–23 were synthesized from the acetyl derivatives 9 and 16 throughout Aldol condensation reaction with the proper aromatic aldehyde using 5% NaOH. The 1H-NMR spectrum reveals the appearance of two doublet peaks in the range of 7.50–8.06 ppm that correspond to the resulting olefinic protons.
These chalcones underwent different cyclization reactions either with hydrazine hydrate in absolute ethanol to afford the pyrazole derivatives 24–29 or with thiourea in the presence of sodium ethoxide to give the pyrimidines 30–35. The most diagnostic aspects in the 1HNMR spectrum that confirmed the pyrazole ring cyclization were the disappearance of the two olefinic protons’ signals and the appearance of a distinguishing singlet that corresponds to pyrazolyl ring proton at 6.5 ppm (Scheme 2).
Compounds 8, 9, 15 and 16 were subjected to bromination in glacial acetic acid to afford compounds 36–39, which were treated with thiourea in the presence of absolute ethanol to give compounds 40–43. IR spectrum was characterized by the presence of NH band at 3,400 cm‒1. In addition, 1HNMR spectra were distinguish by the presence of a singlet signal at 7.11–7.65 ppm, corresponding to the protons of the NH2 group (Scheme 3).
Biological evaluation
Preliminary in vitro cytotoxic screening
In vitro cytotoxicity of all the synthesized imidazolone-derivatives was evaluated against four tumor cell lines: Hela, MCF-7, PC3, and HCT-116, taking Doxorubicin (DOX.) as a control, where IC50 values were determined and listed in Table 1. Compounds 6, 25, 26, and 29 were shown to have the highest anticancer activities in the four cell lines in comparison to DOX. Concerning PC3 cell lines, compound 30 showed a cytotoxic effect compared to that of the standard with IC50 values of 8.15 ± 0.9 μM, while compounds 4 and 18 showed moderate activity with IC50 range of 10.58–11.45 μM. Regarding Hela and HCT-116 cell lines, compound 25 achieved the maximum cytotoxic activity with IC50 values of 7.34 ± 0.5 μM and 4.87 ± 0.3 compared to IC50 values of 5.57 ± 0.4 μM and 5.23 ± 0.2 μM for DOX, respectively. On the other hand, compound 29 depicted the maximum effectiveness in MCF-7 and PC-3 cell lines with IC50 values of 6.27 ± 0.4 μM and 5.31 ± 0.5 μM, respectively, whereas compound 6 reported the least anti-neoplastic activity among the four compounds. Interestingly, compound 29 was approximately twice as potent as Dox in PC3 cell line, while compound 25 was marginally better than the control in the HCT-116 cell line (Fig. 3).
CDK2A inhibition assay
To evaluate the possible mode of action of the synthesized imidazolones as anticancer agents, compounds 6, 25, 26, and 29 were tested for both CDK2A and VEGFR-2 inhibitory activity using sorafenib as a reference drug. All substrates apart from 29 depicted a marginal rise in IC50 compared to sorafenib with compound 26 achieving the best result, followed by compounds 25 and 6. On the other hand, the IC50 of compound 29 skyrocketed up to 3.5 μM making it the least effective (Table 2). These results indicated that CDK2A may be a possible target for the designed hybrids for their antineoplastic activity.
VEGFR-2 enzyme assay
Evaluating the inhibitory effect of compounds 6, 25, 26, and 29 on VEGFR-2 revealed excessively higher IC50 in comparison to sorafenib. IC50 ranged from as low as treble that of sorafenib in compound 6, up to approximately 8 folds in compound 29 (Table 2).
SAR discussion
Several imidazolone hybrids were synthesized and evaluated for their cytotoxicity against four cancer cell lines, namely, Hela, MCF-7, PC3, and HCT-116. Enzyme inhibitory activity against CDK2A as well as VEGFR-2 was also investigated. Two compounds from the first series which embraces furanylmethylene-imidazolone 4–10 showed considerable cytotoxic activity, where the unsubstituted phenyl derivative 4 exhibited moderate activity against PC3 cell lines with an IC50 value of 11.45 ± 1.2 μM, while the 4-methylphenyl derivative 6 showed good and broad-spectrum activity against the tested cell lines. The thiophenyl methylene-imidazolone series 11–17 exhibited weak to no activity. Regarding the imidazolone hybrids containing chalcone moiety 18–23, compound 18 showed moderate activity against PC3 cell lines with an IC50 value of 10.85 ± 1.1 μM. The series that involves the pyrazolyl derivatives 24–29 was the most promising regarding the anticancer activity with three potent compounds 25, 26, and 29, having broad activity against all cell lines. On the other hand, hybrids containing the pyrimidinethione derivatives 30–35 provided compound 30 with anticancer activity compared to that of the standard DOX against PC3 cell lines. The last series 36–43 with the aminothiazole substitution showed low activity.
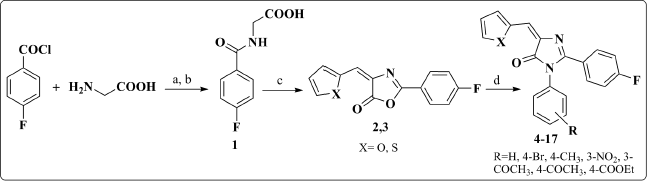 | Scheme 1. Synthesis of compounds 2–17. Reagents and Conditions: (a) 10% NaOH, stirring, 2 hours; (b) conc. HCl; (c) the appropriate heterocyclic aldehyde, NaOAc, glacial acetic acid, reflux; (d) the appropriate aromatic amine, NaOAc/glacial acetic acid, heat/water bath. [Click here to view] |
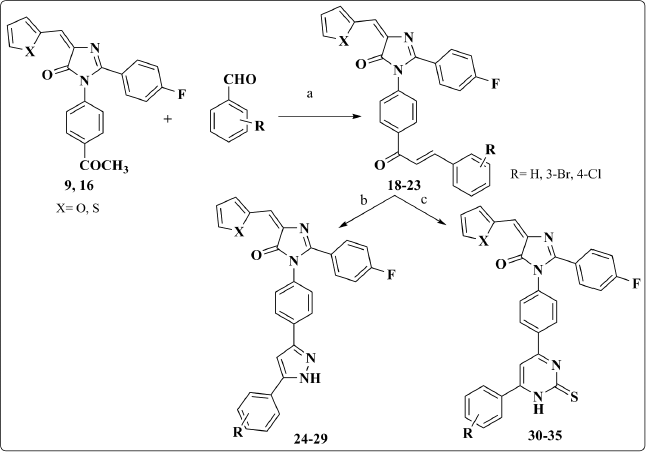 | Scheme 2. Synthesis of compounds 18–35. Reagents and Conditions: (a) 5% ethanolic NaOH, stirring, ice bath, 4h; (b) NH2NH2.H2O, abs. EtOH, reflux; (c) thiourea, NaOEt, reflux. [Click here to view] |
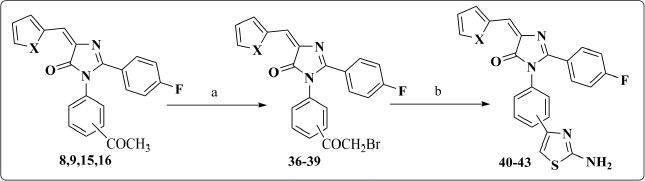 | Scheme 3. Synthesis of compounds 36–43. Reagents and Conditions: (a) Br2, glacial acetic acid, 80°C–90°C; (b) thiourea, abs. EtOH, reflux. [Click here to view] |
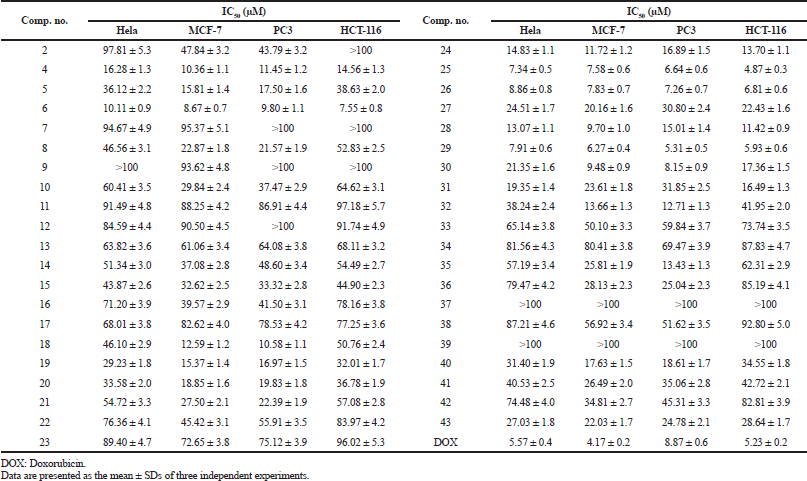 | Table 1. IC50 of the target compounds against Hela, MCF-7, PC3, and HCT-116cell lines. [Click here to view] |
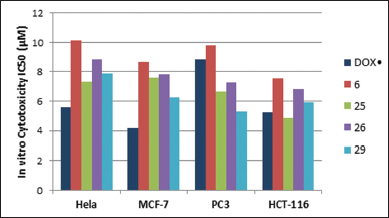 | Figure 3. Cytotoxic activity of compounds 6, 25, 26, and 29 compared to Dox. [Click here to view] |
Concerning the enzyme inhibitory activity against both CDK2A and VEGFR-2, compounds 6, 25, 26, and 29 showed various activities, related to the structural features characterizing them. Ä°nvestigation into the SAR of the target compounds revealed that furanylmethylene-imidazolone hybrids 6, 25, and 26 exhibited considerable enzyme inhibition rather than the thiophenyl methylene-imidazolone hybrid 29, where compound 26 showed an IC50 value of 92 ± 1.57 nM (for VEGFR-2) and 0.66 ± 0.03 μM (for CDK2A) on HCT116 cell line, respectively. Likewise, compound 6 with an IC50 value of 67 ± 0.89 nM (for VEGFR-2) and compound 25 with an IC50 value of 0.83 ± 0.04 μM (for CDK2A) came next. It was observed that compound 6 demonstrated the highest inhibitory effect when compared to other compounds on VEGFR-2 and moderate effect on CDK2A enzyme. On the other hand, compound 29 yielded the lowest inhibitory activity on both tested enzymes. Conversely, when thiophene ring of compound 29 was replaced by furfural ring and the 3-bromophenyl group substituted the 4-chlorophenyl group in compound 25, the inhibitory effect was moderate against CDK2A enzyme. Interestingly, as 3-bromophenyl moiety was replaced by 4-chlorophenyl moiety in the presence of furfural ring in compound 26, the inhibitory effect was demonstrated on both VEGFR-2 and CDK2A enzymes suggesting a powerful anti-cancer effect.
 | Table 2. CDK2A and VEGFR-2 IC50 of compounds 6, 25, 26, and 29 compared to sorafenib. [Click here to view] |
To sum up, compounds 25 and 26 are more likely to produce their effects on the molecular enzymatic level rather than inhibiting cell surface receptors.
CONCLUSION
Compounds 6, 25, 26, and 29 were shown to have the highest anticancer activities in the four cell lines in comparison to DOX. Concerning PC3 cell lines, compound 30 showed a cytotoxic effect comparable to that of the standard. Compounds 6, 25, and 26 depicted a marginal rise in IC50 compared to sorafenib, with compound 26 achieving the best result regarding CDK2A and VEGFR-2 inhibitory activity.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
Ajeesh Kumar A.K, Bodke YD, Gowda AN, Sambasivam G, Bhat KG. Design, synthesis, and evaluation of the anticancer properties of a novel series of imidazolone fused pyrazolo[1,5-a]pyrimidine derivatives. J Heterocyclic Chem, 2017; 54:1904–24. CrossRef
Akhtar J, Khan AA, Ali Z, Haider R, Shahar Yar M. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur J Med Chem, 2017; 125:143–89. CrossRef
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods, 1986; 89:271–7. CrossRef
Dražić T, Vazdar K, Vazdar M, Äaković M, Mikecin AM, Kralj M, Malnar M, Hećimović S, Habuš I. Synthesis of new 2-aminoimidazolones with antiproliferative activity via base promoted amino-β-lactam rearrangement. Tetrahedron, 2015; 71:9202–15. CrossRef
El-Araby M, Omar A, Hassanein HH, El-Helby A-GH, Abdel-Rahman AA. Design, synthesis and in vivo anti-inflammatory activities of 2,4-diaryl-5-4H-imidazolone derivatives. Molecules, 2012; 17:12262. CrossRef
El-Mekabaty A. Erlenmeyer azlactones: synthesis, reactions and biological activity. Int J Modern Org Chem, 2013; 2:40–66. CrossRef
Erlenmeyer E. Ueber die partielle verwandlung der phenyloxyacrylsäure in phenylbrenztraubensäure. Eur J Inorg Chem, 1900; 33:3001–2. CrossRef
Kalyanaraman B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol, 2017; 12:833–42. CrossRef
Kamal A, Ramakrishna G, Raju P, Viswanath A, Ramaiah MJ, Balakishan G, Pal-Bhadra M. Synthesis and anti-cancer activity of chalcone linked imidazolones. Bioorg Med Chem Lett, 2010; 20:4865–9. CrossRef
Kortiwala N, Patel J, Desai VA. Imidazolone and its various biological activities–a review. J Chem Chem Sci, 2016; 6:25–32.
Kumar D, Mariappan G, Husain A, Monga J, Kumar S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab J Chem, 2017; 10:344–50. CrossRef
Mahapatra DK, Bharti SK, Asati V. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem, 2015; 98:69–114. CrossRef
Mai CW, Yaeghoobi M, Abd-Rahman N, Kang YB, Pichika MR. Chalcones with electron-withdrawing and electron-donating substituents: anticancer activity against TRAIL resistant cancer cells, structure-activity relationship analysis and regulation of apoptotic proteins. Eur J Med Chem, 2014; 77:378–87. CrossRef
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods, 1983; 65:55–63. CrossRef
Nasir Abbas Bukhari S, Jasamai M, Jantan I, Ahmad W. Review of methods and various catalysts used for chalcone synthesis. Mini Rev Org Chem, 2013; 10:73–83. CrossRef
Padmaja A, Rajasekhar C, Muralikrishna A, Padmavathi V. Synthesis and antioxidant activity of oxazolyl/thiazolylsulfonylmethyl pyrazoles and isoxazoles. Eur J Med Chem, 2011; 46:5034–8. CrossRef
Premakumari C, Muralikrishna A, Padmaja A, Padmavathi V, Park SJ, Kim T-J, Reddy GD. Synthesis, antimicrobial and anticancer activities of amido sulfonamido methane linked bis heterocycles. Arab J Chem, 2014; 7:385–95. CrossRef
Ramaiah MJ, Pushpavalli SN, Krishna GR, Sarma P, Mukhopadhyay D, Kamal A, Bhadra U, Bhadra MP. Chalcone-imidazolone conjugates induce apoptosis through DNA damage pathway by affecting telomeres. Cancer Cell Int, 2011; 11:11. CrossRef
Rapolu S, Alla M, Bommena VR, Murthy R, Jain N, Bommareddy VR, Bommineni MR. Synthesis and biological screening of 5-(alkyl(1H-indol-3-yl))-2-(substituted)-1,3,4-oxadiazoles as antiproliferative and anti-inflammatory agents. Eur J Med Chem, 2013; 66:91–100. CrossRef
Spano V, Pennati M, Parrino B, Carbone A, Montalbano A, Lopergolo A, Zuco V, Cominetti D, Diana P, Cirrincione G, Zaffaroni N. [1,2]Oxazolo[5,4-e]isoindoles as promising tubulin polymerization inhibitors. Eur J Med Chem, 2016; 124:840–51. CrossRef
Yin S, Tang C, Wang B, Zhang Y, Zhou L, Xue L, Zhang C. Design, synthesis and biological evaluation of novel EGFR/HER2 dual inhibitors bearing a oxazolo[4,5-g]quinazolin-2(1H)-one scaffold. Eur J Med Chem, 2016; 120:26–36. CrossRef
Zhang L, Peng X-M, Damu GLV, Geng R-X, Zhou C-H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med Res Rev, 2014; 34:340–437. CrossRef