
INTRODUCTION
Colorectal cancer (CRC) is a leading cause of cancer-related death in both men and women worldwide (Siegel et al., 2017). Treatment options for CRC depend primarily on the stage of the disease. Standard treatments for CRC are surgery, radiotherapy, chetmotherapeutic drugs, and targeted anticancer drugs. 5-Fluorouracil combined with other chemotherapeutic drugs, such as oxaliplatin and irinotecan, has been the first line chemotherapy for CRC. However, nonselective toxicity for normal cells and acquired drug resistance remain critical challenges of chemotherapy (van der Stok et al., 2017). Therefore, several strategies have been investigated to improve the use of anticancer agents for CRC treatment.
Epidemiological studies have found that nonsteroidal anti-inflammatory drugs (NSAIDs) can decrease CRC-related mortality (Hua et al., 2017). NSAIDs inhibit activities of cyclooxygenases (COXs), including COX-1 and COX-2 isoforms. COX-2 is overexpressed in many types of cancers, including CRC. Overexpression of COX-2 is found in approximately 80% of colon carcinoma (Wu et al., 2003). Prostaglandin E2 (PGE2), a major product of COX-2 catalysis, plays important roles in tumorigenesis and cancer progression by increasing cell survival, proliferation and invasion, stimulating angiogenesis, and inhibiting apoptosis (Greenhough et al., 2009; Sobolewski et al., 2010). Cancer cells overexpressing COX-2 are resistant to many chemotherapeutic agents such as cisplatin, 5-fluorouracil, and paclitaxel (Choi et al., 2011; Ferrandina et al., 2006; Saikawa et al., 2004). NADPH oxidases (NOXs) are major enzymes generating superoxide anion (O2–), which rapidly converts to H2O2. NOX1-dependent ROS is produced during the proliferation and migration of colon cancer cells (Sadok et al., 2008). Celecoxib (CLX; Fig 1), a selective COX-2 inhibitor, is approved by the U.S. Food and Drug Agency to treat patients with familial adenomatous polyposis to prevent colon cancer (Kraus et al., 2013). CLX also demonstrates potent anticancer activity in various cancer cells such as lymphoma, leukemia, breast cancer, pancreatic cancer, and CRC (Grosch et al., 2001; Wang et al., 2017; Wun et al., 2004; Zhang et al., 2006; Zuo et al., 2018). However, long-term use of high-dose CLX can increase the risk of cardiovascular side effects (Kraus et al., 2013). Therefore, an approach of using compounds that have both anticancer and anti-inflammatory activities in combination with CLX for colon cancer treatment is highly promising because these compounds may augment cytotoxicity and decrease side effects associated with CLX.
 | Figure 1. The chemical structure of celecoxib. [Click here to view] |
Cepharanthine (CEP; Fig 2), a biscoclaurine alkaloid isolated from the root of Stephania cepharantha Hayata, is approved by Japanese Ministry of Health for treatment of alopecia areata, leukopenia, and idiopathic thrombocytopenic purpura (Rogosnitzky et al., 2011). Moreover, it demonstrates many pharmacological activities, including antimalarial, anti-HIV-1, antiallergic, anti-inflammatory, and antitumor activities. CEP exerts cytotoxicity against many types of cancer cells by inducing cell cycle arrest and apoptosis, activating ROS production, and downregulating COX-2 expression (Biswas et al., 2006; Chen et al., 2012; Fang et al., 2013; Hua et al., 2015). It may also augment the anticancer activities of several chemotherapeutic agents, such as doxorubicin and paclitaxel (Nakajima et al., 2004; Zhou et al., 2009). Therefore, the present study aimed to determine the combined effects of CEP and CLX and their underlying mechanism(s) in CRC cells.
MATERIALS AND METHODS
Chemicals
CEP was obtained from Abcam. CLX, resazurin, and trypan blue were obtained from Sigma-Aldrich. Other chemicals used were of analytical grade or higher.
Cell culture
Authenticated HT-29 human CRC cells were obtained from the American Type Culture Collection. The cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin at 37°C under a humidified atmosphere of 5% CO2. For cell viability assays, HT-29 cells at low passage were seeded into 96-well plates at 5x104 cells/ml. For all other assays, the cells were seeded into 6-well plates at 5x104 cells/ml.
 | Figure 2. The chemical structure of cepharanthine. [Click here to view] |
Cell viability assay
HT-29 cells were treated with CLX at concentrations of 2.5, 5, 10, 20, and 50 μM, CEP at concentrations of 1.25, 2.5, 5, 10, and 20 μM, or 0.2% dimethyl sulfoxide (DMSO; vehicle control) for 48 hours. Five hours before the end of incubation, 15 ml of 0.05 mg/μl resazurin solution was added into each well. The absorbance of the well contents was measured at 570 and 600 nm using a microplate reading spectrophotometer. IC50 values were calculated using GraphPad Prism 7 software.
Drug interaction analysis
We assessed the interaction of CLX and CEP using a combination index (CI). HT-29 cells were treated with 5, 10, 20, or 40 μM CLX, 1.25 or 2.5 2.5 μM CEP, or their combinations for 48 hours. Viability of the treated cells was measured using a resazurin assay. The CI was determined according to the method described by Chou–Talalay (2010) using the following formula
Dalone1 and Dalone 2: IC30 of the first drug and second drug, respectively.
Dcombination1 and Dcombination2: The concentrations of the first drug and the second drug that gives 30% of cell inhibition, respectively.
CI values were interpreted as follows: CI = 1 was considered to indicate an additive effect, CI < 1 synergism, and CI > 1 antagonism.
Reactive oxygen species assay
The level of intracellular reactive oxygen species (ROS) in HT-29 cells was measured using 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA). After incubating the cells overnight, they were washed with PBS and treated with 100 μl DCFH-DA in Hank’s balanced salts solution for 30 minutes. DCFH-DA was removed and cells were treated with 20 or 40 μM CLX, 1.25 or 2.5 μM CEP, or their combinations for 1 hour. The cells were then lysed with 1% Triton X-100 in 0.3 M NaOH. Subsequently, cell lysates were transferred into 96-well black flat-bottomed polystyrene plates. The fluorescence intensity of the well contents was measured at 485 and 570 nm using a microplate reading fluorescence spectrophotometer.
ELISA for prostaglandin-E2
After incubating HT-29 cells overnight, they were treated with 20 or 40 μM CLX, 1.25 or 2.5 μM CEP, or their combinations for 24 hours. Supernatants were collected and stored at -20°C until they were assayed for prostaglandin-E2 (PGE2). PGE2 levels in the cells were measured using a Prostaglandin E2 Human ELISA Kit (Invitrogen–Thermo Scientific) according to the manufacturer’s instructions.
Flow cytometric analysis of cell cycle stage
After incubating HT-29 cells overnight, they were treated with 20 or 40 μM CLX, 1.25 or 2.5 μM CEP, or their combinations for 48 hours. Cells were collected after trypsinization, washed with PBS, and fixed with 70% ethanol at –20°C for 20 minutes. Cells were then washed with PBS, resuspended in 500 μl PBS, and incubated with 20 μg RNase at room temperature for 1 hour. Subsequently, cells were stained with 5 μl propidium iodide (Thermo Scientific) for 15 minutes. The DNA content of 10,000 cells/sample was determined using a flow cytometer.
Cell apoptosis analysis
After incubating HT-29 cells overnight, they were treated with 20 or 40 μM CLX, 1.25 or 2.5 μM CEP, or their combinations for 24 hours. Cells were collected, washed with PBS, and resuspended in 100 μM assay buffer specified for the Annexin V-FITC Apoptosis Detection Kit (Life Technologies). Briefly, following the protocol specified by the manufacturer, cells were stained with 1 μl fluorescein isothiocyanate-labeled Annexin V for 15 minutes at room temperature followed by 1 μl propidium iodide for 5 minutes on ice. Four populations of stained cells, including viable cells (Annexin V–, PI–), early apoptotic cells (Annexin V+, PI–), late apoptotic cells (Annexin V+, PI+), and necrotic cells (Annexin V–, PI+) were counted using a flow cytometer.
qPCR analysis
After incubating HT-29 cells overnight, they were treated with 20 or 40 μM CLX, 1.25 or 2.5 μM of CEP, or their combinations for 24 hours. Total RNA was isolated using TRIzol reagent according to manufacturer’s instructions (Life Technologies). The RNA was transcribed to cDNA using an ImProm-II Reverse Transcription System (Promega). The genes of interest were amplified using SYBR Green I with specific primers listed in Table 1. Real-time qPCR reactions were conducted using a StepOnePlus Real-Time PCR System (Applied Biosystems) using the following conditions: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. Gene expression was calculated using the 2-DDCT method.
Statistical analysis
The data are presented as mean ± standard error of the mean (SEM) of three independent experiments. Groups were compared using a one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test. Differences with p < 0.05 were considered significant.
| Table 1. The sequencing primers used for qPCR. [Click here to view] |

RESULTS
Celecoxib with cepharanthine displayed synergistic cytotoxicity for HT-29 cells
Initially, we assessed the individual effects of CLX or CEP on the viability of HT-29 CRC cells using a resazurin assay. Treatment with CLX at 40 or 50 μM significantly decreased the viability of HT-29 cells with an IC50 > 50 μM (Fig 3A). In contrast to CLX, CEP decreased the viability of HT-29 in a concentration-dependent manner with an IC50 of 5.22 ± 0.28 μM (Fig 3B). We then determined the effect of CLX at 5, 10, 20, or 40 μM combined with CEP at 1.25 or 2.5 μM, concentrations less than their IC50s, respectively, on HT-29 cell viability. As shown in Fig 1C, only 40 μM CLX induced cell death significantly compared with the vehicle control. However, treatment with 5, 10, 20, or 40 μM CLX combined with 1.25 μM CEP resulted in significant cell death compared with the vehicle control and each drug alone. In contrast to 1.25 μM CEP, we found that 2.5 μM CEP was cytotoxic for HT-29 cells, but cytotoxicity that enhanced significantly more than that for each drug alone was found after treatment of HT-29 cells with 20 or 40 μM CLX combined with 2.5 μM CEP.
The effect of the CLX–CEP combination on the viability of HT-29 cells was also interpreted as the CI. The IC30 values of CLX (48.18 ± 1.53 μM) and CEP (3.08 ± 0.22 μM) were used to calculate the CI for each combination. The results shown in Table 2 indicate the CIs of the combinations were <1. Thus, the combinations had a nearly additive effect (CI = 0.964) or from moderate to strong synergistic effects (CI range: 0.424–0.777) on the viability of HT-29 cells. Combinations of 20 or 40 μM CLX with 1.25 or 2.5 μM CEP, which had a CI range of 0.424–0.694, were chosen for further investigation.
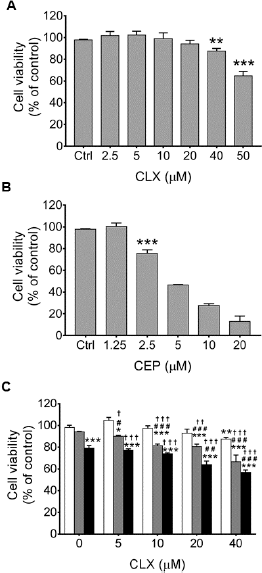 | Figure 3. Effects of celecoxib, or cepharanthine, or their combination on HT-29 cell viability. Cells were treated with (A) celecoxib (CLX) at 2.5, 5, 10, 20, 40, or 50 μM, (B) cepharanthine (CEP) at 1.25, 2.5, 5, 10, or 20 μM, or (C) combinations of 5, 10, 20, or 40 μM of CLX alone (unfilled bars) with 1.25 (gray bars) or 2.5 μM (black bars) CEP for 48 h. The viability of the treated cells was assayed using resazurin. The data are presented as mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. CEP alone; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. CLX alone. [Click here to view] |
Effects of celecoxib and cepharanthine combined on COX-2 mRNA levels and PGE
CLX (40 μM) significantly decreased COX-2 mRNA levels in HT-29 cells by approximately 45% compared with the vehicle control (Fig 4A). In contrast, neither 1.25 μM nor 2.5 μM of CEP affected the level of COX-2 mRNA in HT-29 cells. It is noteworthy that COX-2 mRNA levels in HT-29 cells treated with the drug combinations were no different from those in cells treated with the vehicle control or either drug alone. CLX (20 or 40 μM) significantly decreased PGE2 levels in HT-29 cells to 73% and 60% of the level produced after treatment with vehicle control, respectively (Fig 4B). Similarly, we found that CEP at 2.5 μM significantly decreased PGE2 production by HT-29 cells compared with the level produced after treatment with vehicle control (Fig 4B). Although 1.25 μM CEP had no detectable effect on PGE2 production by HT-29 cells, the combination of 40 μM CLX and 1.25 μM CEP significantly decreased the level of PGE2 production by the cells compared with the level produced after treatment with CLX alone. In contrast to the low concentration of CEP, 20 or 40 μM CLX combined with 2.5 μM CEP had no detectable effect on PGE2 production by HT-29 cells compared with the effect of treatment of HT-29 cells with either agent individually.
.png) | Table 2. Combination index values for CLX–CEP. [Click here to view] |
 | Figure 4. Effects of treatment of HT-29 cells with celecoxib, or cepharanthine, or their combination on COX-2 mRNA levels and PGE2 production. Cells were treated with 20 or 40 μM of celecoxib (CLX) alone (unfilled bars), or 1.25 μM (gray bars) or 2.5 μM (black bars) of cepharanthine (CEP), or their combination for 24 h. Levels of (A) COX-2 mRNA were determined by qPCR and (B) PGE2 production by competitive ELISA. The data are presented as mean ± SEM from three independent experiments. *P < 0.01, **P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; ##P < 0.01, ###P < 0.001 vs. CEP alone. [Click here to view] |
 | Figure 5. Effects of celecoxib, or cepharanthine, or their combination on NOX-1 mRNA levels and ROS production in HT-29 cells. (A) Cells were treated with 20 or 40 μM celecoxib (CLX) alone (unfilled bars), or 1.25 μM (gray bars) or 2.5 μM (black bars) of cepharanthine (CEP), or their combinations for 24 h. Levels of NOX-1 mRNA were determined by qPCR. (B) Cells were pretreated with 50 μM DCFH-DA for 30 min and treated with 20 or 40 μM CLX, 1.25 μM or 2.5 μM CEP, or their combinations for 1 h. The fluorescence of DCF product was determined using a microplate reading fluorescence spectrophotometer. The data are presented as mean ± SEM from three independent experiments. *P < 0.01, **P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; ##P < 0.01, ###P < 0.001 vs. CEP alone. [Click here to view] |
Effects of celecoxib and cepharanthine combined on NOX1 mRNA levels and ROS production by HT-29 cells
CLX dramatically downregulated NOX1 transcription in a concentration-dependent manner (Fig 5A). After treatment of HT-29 cells with 20 or 40 μM CLX, cellular NOX1 mRNA levels were decreased by approximately 60% or 80%, respectively, compared with those after treatment with the vehicle control. In contrast, we found that neither 1.25 nor 2.5 μM CEP altered NOX1 mRNA levels (Fig 5A). Moreover, CEP alone had no effect on CLX-induced NOX1 downregulation (Fig 5A). NOX1 mRNA levels in HT-29 cells treated with CLX combined with CEP were not different from those in cells treated with CLX alone. Neither CLX nor CEP altered ROS production by HT-29 cells compared with the vehicle control (Fig 5B). However, we found that 40 μM CLX combined with 1.25 μM CEP and 20 μM CLX combined with 2.5 μM CEP decreased ROS levels significantly compared with the levels after treatment with the vehicle control. Furthermore, 40 μM CLX combined with 2.5 μM CEP decreased ROS levels significantly compared with the levels after treatment with either the vehicle control or CEP alone (Fig 5B).
 | Figure 6. Effects of celecoxib, or cepharanthine, or their combination on cell cycle progression in HT-29 cells. Cells were treated with 20 or 40 μM celecoxib (CLX), 1.25 μM or 2.5 μM cepharanthine (CEP), or their combinations for 48 h. The treated cells were fixed and stained with PI and analyzed using a flow cytometer to determine the percentage of the treated cells in each phase of the cell cycle. The data are presented as mean ± SEM from three independent experiments. *P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; #P < 0.05, ##P < 0.01, ### P < 0.001 vs. CEP alone; ††P < 0.01, †††P < 0.001 vs. CLX alone. [Click here to view] |
Celecoxib combined with cepharanthine induced HT-29 cell cycle arrest synergistically
Flow cytometry indicated that treatment of HT-29 cells with 1.25 μM CEP or 40 μM CLX individually caused significant cell accumulation at the G2/M or G1 phase, respectively (Fig 6). Remarkably, treatment of HT-29 cells with 20 μM or 40 μM CLX combined with 1.25 μM CEP caused significant cell accumulation at the G1 phase compared with that after treatment with either drug alone. In contrast to 1.25 μM CEP, 2.5 μM CEP blocked cell cycle progression at the G1 phase significantly. Accumulation of cells in the G1 phase was also detected after treatment of HT-29 cells with 20 μM CLX combined with 2.5 μM CEP or 40 μM CLX combined with 2.5 μM CEP. It is noteworthy that a significant increase of cells in the G1 phase was associated with a significant reduction of cells in the S and G2/M phases.
Treatment of HT-29 cells with either CLX or CEP alone downregulated cyclin A2 mRNA levels significantly in a concentration-dependent manner (Fig 7A). Remarkably, treatment of HT-29 cells with 40 μM CLX combined with 1.25 or 2.5 μM CEP downregulated cyclin A2 mRNA levels significantly compared with the levels seen after treatment with either drug alone. In contrast, treatment with the same concentrations of either CLX or CEP alone or their combination did not apparently affect cyclin D1 mRNA levels (Fig 7B). Although we observed that treatment of HT-29 cells with 20 or 40 μM CLX alone decreased cyclin E mRNA levels significantly compared with levels found after treatment with the vehicle control, there was no synergistic effect of CLX and CEP on cyclin E1 mRNA levels (Fig 7C). Cyclin E1 mRNA levels in cells treated with CLX alone were not different from those in cells treated with CLX and CEP combined. In contrast to cyclins, a synergistic effect of CLX and CEP on increased levels of mRNA for p21 was found (Fig 7D). Only CLX at 40 μM increased p21 mRNA levels significantly, but all combinations increased p21 mRNA levels. In the presence of 1.25 μM CEP, treatment with either 20 or 40 μM CLX increased p21 mRNA levels by approximately 4 and 9 times, respectively, the levels seen after treatment with vehicle control. In the presence of 2.5 μM CEP, treatment with either 20 or 40 μM CLX also increased p21 mRNA levels significantly. Taken together, these results suggest that downregulation of CCNA2, the gene for cyclin A2, and upregulation of CDKN1A, the gene for p21 are targets of the synergistic interaction between CLX and CEP in the induction of cell cycle arrest in HT-29 cells.
 | Figure 7. Effects of celecoxib, or cepharanthine, or their combination on mRNA levels of cell cycle regulators in HT-29 cells. Cells were treated with 20 or 40 μM celecoxib (CLX) alone (unfilled bars), or 1.25 μM (gray bars) or 2.5 μM (black bars) of cepharanthine (CEP), or their combinations for 24 h. The levels of mRNA for (A) cyclin A, (B) cyclin D, (C) cyclin E, and (D) p21 in the treated cells was determined using qPCR. The data are presented as mean ± SEM from three independent experiments. *P < 0.01, **P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; #P < 0.05, ##P < 0.01 vs. CEP alone; †P < 0.05, ††P < 0.001 vs. CLX alone. [Click here to view] |
 | Figure 8. Effects of celecoxib, or cepharanthine, or their combination on apoptosis in HT-29 cells. Cells were treated with 20 or 40 μM celecoxib (CLX), 1.25 μM or 2.5 μM cepharanthine (CEP), or their combinations for 24 h. The treated cells were fixed, and stained with Annexin V-FIT-C/PI and analyzed using a flow cytometer. The data are presented as mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. 0.2% DMSO vehicle control; ##P < 0.01, ###P < 0.001 vs. CEP alone; ††P < 0.01, †††P < 0.001 vs. CLX alone. [Click here to view] |
Celecoxib and cepharanthine had a synergistic effect on the apoptosis of HT-29 cells
Treatment with 20 or 40 μM CLX alone or 1.25 μM CEP alone did not induce significant apoptosis of HT-29 cells (Fig 8). However, treatment of the cells with their combinations induced apoptosis significantly compared with treatment with either drug alone. Treatment of HT-29 cells with 20 μM CLX combined with 1.25 μM CEP or 40 μM CLX combined with 1.25 μM CEP increased numbers of late apoptotic cells, respectively, by 10% or 15% of the number induced by treatment with vehicle control. Similarly, treatment of the cells with 20 or 40 μM of CLX combined with 2.5 μM CEP triggered apoptosis significantly compared with apoptosis after treatment with either drug alone. In the presence of 2.5 μM CEP, 20 or 40 μM CLX increased numbers of late apoptotic cells, respectively, by approximately 2 and 2.5 times the number after treatment with the vehicle control. Neither CLX nor CEP treatment of HT-29 cells altered BAX mRNA levels, whereas all combinations increased BAX mRNA levels significantly compared with the expression after treatment with vehicle control (Fig 9A). Remarkably, treatment with 20 μM CLX combined with 2.5 μM CEP increased BAX mRNA levels by approximately 2.1 and 1.7 times those seen after treatment with the vehicle control and CEP alone, respectively. Similarly, treatment of the cells with 40 μM CLX combined with 2.5 μM CEP increased BAX mRNA levels significantly compared with the levels found after treatment with vehicle control or either drug alone. In contrast to BAX, only treatment of the cells with 20 μM CLX combined with 2.5 μM CEP increased BAK mRNA levels significantly compared with the vehicle control and each drug alone. For antiapoptotic proteins, we found that Bcl-xL mRNA levels were unaffected by treatment with a single drug. However, a significant increase in Bcl-xL mRNA levels, compared with those found after treatment of HT-29 cells with vehicle control or CLX alone, were detected after treatment of the cells with 20 μM CLX combined with 1.25 μM CEP, 20 μM CLX combined with 2.5 μM CEP, or 40 μM CLX combined with 2.5 μM CEP. Notably, we did not observe synergistic effects of CLX and CEP on levels of Bcl-2 mRNA. A significant decrease in Bcl-2 mRNA level (65% of that after treatment of HT-29 cells with vehicle control) was detected in cells treated with 2.5 μM CEP, but there was no significant difference in Bcl-2 mRNA levels between cells treated with 2.5 μM CEP, or with 2.5 μM CEP combined with 20 or 40 μM CLX.
 | Figure 9. Effects of celecoxib, or cepharanthine, or their combination on mRNA levels of Bcl-2 family proteins in HT-29 cells. Cells were treated with 20 or 40 μM of celecoxib (CLX) alone (unfilled bars), or 1.25 μM (gray bars) or 2.5 μM (black bars) of cepharanthine (CEP), or their combinations for 24 h. The levels of mRNA for (A) BAX, (B) BAK, (C) Bcl-xL, and (D) Bcl-2 in the treated cells was determined by quantitative real time RT-PCR. The data are presented as mean ± SEM from three independent experiments. *P < 0.01, **P < 0.01 vs. 0.2% DMSO vehicle control; #P < 0.05 vs. CEP alone; †P < 0.05, ††P < 0.01 vs. CLX alone. [Click here to view] |
DISCUSSION
In the present study, we found that CLX or CEP alone decreased the viability of HT-29 cells with IC50 values of >50 and 5.22 μM, respectively. Remarkably, the combination of CLX and CEP, at their sub-IC50 concentrations, synergistically had cytotoxic activity on HT-29 cells. The cytotoxic activity of CLX is strongly associated with reduced PGE2 (Sobolewski et al., 2010) and ROS production (Sullivan et al., 2014). ROS and NOX-1 control COX-2 expression and PGE2 production (Lin et al., 2011; Shimizu et al., 2015). We found that the drug combinations decreased PGE2 and ROS production and downregulated NOX1 transcription in HT-29 cells. Treatment of the cells with CLX alone decreased PGE2 production and downregulated NOX1 transcription, but did not have an apparent effect on ROS production. These findings were consistent with the findings of Banskota et al. (2015) that CLX did not inhibit tetradecanoylphorbol-13-acetate-induced ROS in HT-29 cells. Although CEP suppressed expression of PTGS2, the gene for COX-2, and stimulated ROS production in other studies (Fang et al., 2013; Hua et al., 2015; Rattanawong et al., 2018), CEP did not have any apparent effect on these variables at the concentrations used in the present study. This apparent inconsistency may be a consequence of the different concentrations of CEP used in our experiments. Because we found no synergism between CLX and CEP on PTGS2 or NOX-1 transcription, nor PGE2 or ROS production, it is possible that the synergistic activity of CLX and CEP, at least at their sub-IC50 concentrations, maybe COX-2 and ROS-independent. Our present findings are consistent with others that showed CLX to potentiate the cytotoxic effect of cisplatin in vulvar cancer cells, regardless of COX-2 expression (Kim et al., 2009).
The present study found that treatment of HT-29 cells with combinations of 20 or 40 μM of CLX with 1.25 or 2.5 μM of CEP increased cell accumulation at the G1 phase significantly compared with the effect of cell treatment with either CLX or CEP alone. Moreover, our mechanistic studies found that compared with vehicle controls, these combinations decreased significantly the levels of mRNA for cyclin E, which activates CDK2 to form a CDK2-cyclin E complex in the late G1 phase of the cell cycle. Compared with treatment by each drug alone, cell treatment with their combinations also dramatically decreased the levels of mRNA for cyclin A, which activates CDK2 and CDK1 in S phase and G2 phase, and increased the levels of p21 mRNA. Notably, a single treatment of HT-29 cells with CLX alone also increased the levels of p21 mRNA and decreased the levels of mRNA for cyclin E and cyclin A, whereas CEP at sub-IC50 concentration only decreased the levels of mRNA for cyclin A. CLX and CEP have been shown to arrest cell cycle progression of not only CRC cells but also several other cancer cell lines. CLX was shown to arrest colon cancer cells at G1 phase (Buecher et al., 2003; Grosch et al., 2001) and chronic myeloid leukemia cells at G1/S phase (Zhang et al., 2006). CLX also upregulated many cyclin-dependent kinase inhibitors, including p21, p16, and 27, and downregulated cyclin A and cyclin B1 expression in CRC cells (Peng et al., 2004). Similarly, CEP induces cell cycle arrest at G1 phase in adenosquamous carcinoma cells, myeloma cells, and CRC cells by upregulating p21 and downregulating several cyclins (Harada et al., 2001; Kikukawa et al., 2008; Rattanawong et al., 2018). Therefore, more pronounced effects of the combinations on cell accumulation at G1 phase, upregulation of p21, and downregulation of cyclin A2 are likely and may be associated with the synergistic cytotoxic effects of individual drugs on HT-29 cells.
In the present study, we found that at sub-IC50 concentrations, a single treatment with CLX or CEP alone did not have apoptosis-inducing effects on HT-29 cells after 24 hours. However, combinations increased the numbers of late apoptotic cells significantly, suggesting that CLX combined with CEP induced HT-29 apoptosis synergistically. Our present findings demonstrated that combinations increased BAX mRNA levels and decreased Bcl-xL mRNA levels significantly. Furthermore, combinations of 20 or 40 μM of CLX with 2.5 μM CEP downregulated transcription of BCL2 moderately, whereas a combination of 40 μM CLX with 2.5 μM CEP upregulated BAX transcription significantly compared with the effect of cell treatment with each drug alone. Our results were consistent with demonstrations that CLX and CEP induced the apoptosis of cancer cells by modulating the expression of Bcl-2 protein family members. CLX upregulates BAX expression in breast cancer cells (Kern et al., 2006), whereas CEP upregulates expression of BAX and downregulates expression of Bcl-2 in non-small cell lung cancer cells (Hua et al., 2015). Changes in the levels of cell cycle regulators also induce apoptosis. Binding of p21 with cyclin A/CDK2 complex leads to activation of caspase 3, an executioner caspase in the apoptotic pathway (Gartel et al., 2002). The upregulation of p21 also correlates with induction of expression of the proapoptotic protein BAX. Moreover, the upregulation of p21 is associated with cell cycle arrest and apoptosis in p53-wild type and p53-mutant cancer cells (Vousden et al., 2002). Genistein, an isoflavonoid, induced p53 mutant breast cancer MDA-MB-231 cells to undergo apoptosis by inducing p21 and BAX expression (Li et al., 1999). Thus, it is possible that, in addition to Bcl-2 family proteins, the effects of the drug combination to induce apoptosis in p53 mutant HT-29 cells may also be mediated through modulation of cell cycle regulators. However, it should be noted that only mRNA and not protein expression has been tested in the present study and protein expression may be different to the relative level of mRNA expression for various reasons. Many variables, including mRNA stability, translation rate, and protein degradation influence the steady-state protein concentration (Maier et al., 2009). Investigating the effect of CLX and CEP combinations on protein expression of cell cycle regulators and Bcl-2 family members may add support to the present findings. Moreover, the present study was conducted in just one cell line. Replication of the present study in various cell lines would support more general applicability of our findings.
CONCLUSION
Treatment of HT-29 cells with CLX and CEP combined inhibited cell proliferation synergistically via cell cycle arrest and apoptosis. The synergism of the drugs was associated with increases in the level of mRNA for p21and BAX and decreases in the levels of mRNA for cyclin A2 and Bcl-xL. Our present findings warrant using combinations of CLX and CEP to determine potential synergistic antitumor effects in animal models, which might suggest a combination is a promising approach to CRC treatment in humans.
ACKNOWLEDGMENTS
This study was supported by the Rathchadaphisek Somphot Fund, Chulalongkorn University, Thailand, grant number 2560-029 (WL) and Special Task Force for Activating Research Ratchadaphiseksomphot Endowment Fund, grant number GSTAR 59-005-30-001.
CONFLICT OF INTEREST
None of the authors declare any potential conflict of interest.
REFERENCES
Banskota S, Regmi SC, Kim JA. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol Cancer, 2015; 14:123. CrossRef
Biswas KK, Tancharoen S, Sarker KP, Kawahara K, Hashiguchi T, Maruyama I. Cepharanthine triggers apoptosis in a human hepatocellular carcinoma cell line (HuH-7) through the activation of JNK1/2 and the downregulation of Akt. FEBS Lett, 2006; 580:703–10. CrossRef
Buecher B, Broquet A, Bouancheau D, Heymann MF, Jany A, Denis MG, Bonnet C, Galmiche JP, Blottiere HM. Molecular mechanisms involved in the antiproliferative effect of two COX-2 inhibitors, nimesulide and NS-398, on colorectal cancer cell lines. Dig Liver Dis, 2003; 35:557–65. CrossRef
Chen Z, Huang C, Yang YL, Ding Y, Ou-Yang HQ, Zhang YY, Xu M. Inhibition of the STAT3 signaling pathway is involved in the antitumor activity of cepharanthine in SaOS2 cells. Acta Pharmacol Sin, 2012; 33:101–8. CrossRef
Choi S, Ku JL. Resistance of colorectal cancer cells to radiation and 5-FU is associated with MELK expression. Biochem Biophys Res Commun, 2011; 412:207–13. CrossRef
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res, 2010; 70:440–6. CrossRef
Fang ZH, Li YJ, Chen Z, Wang JJ, Zhu LH. Inhibition of signal transducer and activator of transcription 3 and cyclooxygenase-2 is involved in radiosensitization of cepharanthine in HeLa cells. Int J Gynecol Cancer, 2013; 23:608–14. CrossRef
Ferrandina G, Ranelletti FO, Martinelli E, Paglia A, Zannoni GF, Scambia G. Cyclo-oxygenase-2 (Cox-2) expression and resistance to platinum versus platinum/paclitaxel containing chemotherapy in advanced ovarian cancer. BMC Cancer, 2006; 6:182. CrossRef
Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther, 2002; 1:639–49.
Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis, 2009; 30:377–86. CrossRef
Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J, 2001; 15:2742–4. CrossRef
Harada K, Bando T, Yoshida H, Sato M. Characteristics of antitumour activity of cepharanthin against a human adenosquamous cell carcinoma cell line. Oral Oncol, 2001; 37:643–51. CrossRef
Hua P, Sun M, Zhang G, Zhang Y, Tian X, Li X, Cui R, Zhang X. Cepharanthine induces apoptosis through reactive oxygen species and mitochondrial dysfunction in human non-small-cell lung cancer cells. Biochem Biophys Res Commun, 2015; 460:136–42. CrossRef
Hua X, Phipps AI, Burnett-Hartman AN, Adams SV, Hardikar S, Cohen SA, Kocarnik JM, Ahnen DJ, Lindor NM, Baron JA, Newcomb PA. Timing of aspirin and other nonsteroidal anti-inflammatory drug use among patients with colorectal cancer in relation to tumor markers and survival. J Clin Oncol, 2017; 35:2806–13. CrossRef
Kern MA, Haugg AM, Koch AF, Schilling T, Breuhahn K, Walczak H, Fleischer B, Trautwein C, Michalski C, Schulze-Bergkamen H, Friess H, Stremmel W, Krammer PH, Schirmacher P, Muller M. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res, 2006; 66:7059–66. CrossRef
Kikukawa Y, Okuno Y, Tatetsu H, Nakamura M, Harada N, Ueno S, Kamizaki Y, Mitsuya H, Hata H. Induction of cell cycle arrest and apoptosis in myeloma cells by cepharanthine, a biscoclaurine alkaloid. Int J Oncol, 2008; 33:807–14.
Kim SH, Kim SH, Song YC, Song YS. Celecoxib potentiates the anticancer effect of cisplatin on vulvar cancer cells independently of cyclooxygenase. Ann N Y Acad Sci, 2009; 1171:635–41. CrossRef
Kraus S, Naumov I, Arber N. COX-2 active agents in the chemoprevention of colorectal cancer. Recent Results Cancer Res, 2013; 191:95–103. CrossRef
Li Y, Upadhyay S, Bhuiyan M, Sarkar FH. Induction of apoptosis in breast cancer cells MDA-MB-231 by genistein. Oncogene, 1999; 18:3166–72. CrossRef
Lin WN, Lin CC, Cheng HY, Yang CM. Regulation of cyclooxygenase-2 and cytosolic phospholipase A2 gene expression by lipopolysaccharide through the RNA-binding protein HuR: involvement of NADPH oxidase, reactive oxygen species and mitogen-activated protein kinases. Br J Pharmacol, 2011; 163:1691–706. CrossRef
Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett, 2009; 583:3966–73. CrossRef
Nakajima A, Yamamoto Y, Taura K, Hata K, Fukumoto M, Uchinami H, Yonezawa K, Yamaoka Y. Beneficial effect of cepharanthine on overcoming drug-resistance of hepatocellular carcinoma. Int J Oncol, 2004; 24:635–45. CrossRef
Peng J, Zhang GY, Xiao ZQ. Effects of celecoxib on the proliferation and apoptosis of human colorectal cancer cell line HT-29. Zhong Nan Da Xue Xue Bao Yi Xue Ban, 2004; 29:261–5.
Rattanawong A, Payon V, Limpanasittikul W, Boonkrai C, Mutirangura A, Wonganan P. Cepharanthine exhibits a potent anticancer activity in p53-mutated colorectal cancer cells through upregulation of p21Waf1/Cip1. Oncol Rep, 2018; 39:227–38. CrossRef
Rogosnitzky M, Danks R. Therapeutic potential of the biscoclaurine alkaloid, cepharanthine, for a range of clinical conditions. Pharmacol Rep, 2011; 63:337–47. CrossRef
Sadok A, Bourgarel-Rey V, Gattacceca F, Penel C, Lehmann M, Kovacic H. Nox1-dependent superoxide production controls colon adenocarcinoma cell migration. Biochim Biophys Acta, 2008; 1783:23–33. CrossRef
Saikawa Y, Sugiura T, Toriumi F, Kubota T, Suganuma K, Isshiki S, Otani Y, Kumai K, Kitajima M. Cyclooxygenase-2 gene induction causes CDDP resistance in colon cancer cell line, HCT-15. Anticancer Res, 2004; 24:2723–8.
Shimizu S, Ishigamori R, Fujii G, Takahashi M, Onuma W, Terasaki M, Yano T, Mutoh M. Involvement of NADPH oxidases in suppression of cyclooxygenase-2 promoter-dependent transcriptional activities by sesamol. J Clin Biochem Nutr, 2015; 56:118–22. CrossRef
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, Jemal A. Colorectal cancer statistics, 2017. CA Cancer J Clin, 2017; 67:177–93. CrossRef
Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol, 2010; 2010:215158. CrossRef
Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab, 2014; 2:17. CrossRef
van der Stok EP, Spaander MCW, Grunhagen DJ, Verhoef C, Kuipers EJ. Surveillance after curative treatment for colorectal cancer. Nat Rev Clin Oncol, 2017; 14:297–315. CrossRef
Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer, 2002; 2:594–604. CrossRef
Wang G, Li J, Zhang L, Huang S, Zhao X, Zhao X. Celecoxib induced apoptosis against different breast cancer cell lines by down-regulated NF-kappaB pathway. Biochem Biophys Res Commun, 2017; 490:969–76. CrossRef
Wu AW, Gu J, Ji JF, Li ZF, Xu GW. Role of COX-2 in carcinogenesis of colorectal cancer and its relationship with tumor biological characteristics and patients' prognosis. World J Gastroenterol, 2003; 9:1990–4. CrossRef
Wun T, McKnight H, Tuscano JM. Increased cyclooxygenase-2 (COX-2): a potential role in the pathogenesis of lymphoma. Leuk Res, 2004; 28:179–90. CrossRef
Zhang GS, Liu DS, Dai CW, Li RJ. Antitumor effects of celecoxib on K562 leukemia cells are mediated by cell-cycle arrest, caspase-3 activation, and downregulation of Cox-2 expression and are synergistic with hydroxyurea or imatinib. Am J Hematol, 2006; 81:242–55. CrossRef
Zhou Y, Hopper-Borge E, Shen T, Huang XC, Shi Z, Kuang YH, Furukawa T, Akiyama S, Peng XX, Ashby CR, Jr., Chen X, Kruh GD, Chen ZS. Cepharanthine is a potent reversal agent for MRP7(ABCC10)-mediated multidrug resistance. Biochem Pharmacol, 2009; 77:993–1001. CrossRef
Zuo C, Hong Y, Qiu X, Yang D, Liu N, Sheng X, Zhou K, Tang B, Xiong S, Ma M, Liu Z. Celecoxib suppresses proliferation and metastasis of pancreatic cancer cells by down-regulating STAT3 / NF-kB and L1CAM activities. Pancreatology, 2018; 18:328–33. CrossRef