
INTRODUCTION
Endophytic fungi are defined as microbes that live colonially in various tissues of plants, such as roots, stems, leaves, tubers, fruits, and flowers (Müller, 2015; Stone et al., 2004; Wei et al., 2016; Zhang et al., 2006). In general, the endophytic fungi do not cause any negative effects during life in host plant tissues (Gao et al., 2005; Kyekyeku et al., 2017). There is a mutually beneficial interaction between endophytic fungi and its host plants. Plants supply nutrients for fungal growth and fungi will produce secondary metabolites to protect the host plants from attacks (Kogel et al., 2006). Although the number of endophytic species is estimated to be more than 1 million, nevertheless the phytochemical data of these microbes in the literature are still limited (Guo et al., 2008; Jia et al., 2016). Therefore, many species of endophytic fungi have not been evaluated chemically.
Artocarpus, the high tree, is an important genus in the mulberry family and fig family, Moraceae. This plant has been widely used as traditional medicine, including anti-inflammation, antimalarial, and remedy to cure fever and diarrhea (Jagtap, 2010; Septama and Panichayupakaranant, 2018). Research on the phytochemical constituents of Artocarpus in our laboratory showed that the main secondary metabolites in the plants of this genus are prenylated phenolic compounds such as flavonoids, stilbenes, and 2-arylbenzofurans with a variety of biological activities such as anti-cancer, antimalarial, antimicrobial, cytotoxic, and antioxidant (Achmad et al., 1996; Hakim et al., 1999; 2002; 2006; Syah et al., 2001; 2004; Mustapha et al., 2009; 2010). To continue our work on the chemical constituents of Artocarpus, we investigated the endophytic fungi from this plant. From this study, we will know the diversity of structures and bioactivity of secondary metabolites from fungi isolated from Artocarpus. An endophytic fungus, identified as HF 12440, was isolated from the stem of Artocarpus heterophyllus. This fungal genus has been shown to produce lactones, phenylpropanoids, alkaloids, terpenoids, quinones, peptides, and xanthones with important bioactivities (Kuang et al., 2016; Xia et al., 2016; Xiao et al., 2017; Xu et al., 2010). In the present study, we report four compounds (1–4) from the liquid media of Pestalotiopsis microspora HF 12440 and their cytotoxic activities against P388 cells. In fact, this is the first reported study about isolation, structure elucidation, and bioactivity test of the secondary metabolites from P. microspora HF 12440 living in A. heterophyllus.
MATERIAL AND METHODS
General experimental procedures
Potato dextrose agar (PDA) and potato dextrose broth (PDB) (Himedia, Mumbai, India) were used for inoculation and cultivation of endophytic fungi. Silica gel 60 G, silica gel Kieselgel 60 (0.063–0.200 mm), silica gel 60 PF254, and silica gel 60 F254 (Merck, Darmstadt, Germany) were used for vacuum liquid chromatography, column chromatography, radial chromatography, and thin layer chromatography, respectively. NMR spectroscopic data were recorded at 500 MHz for 1H and 125 MHz for 13C on Agilent Varian with CDCl3 and (CD3)2CO as the solvents and tetramethylsilane (TMS) as the internal standard (Agilent Technologies, Santa Clara, CA). MS data were recorded using a Bruker spectrometer (Bruker Corporation, Billerica, MA). Optical rotation was measured on an Autopol IV polarimeter (Rudolph Research Analytical, New Jersey).
Isolation of endophytic fungus
The stem of A. heterophyllus was collected from the garden of School of Pharmacy in Bandung Institute of Technology, Indonesia in December 2016. The stem of A. heterophyllus was sterilized with ethanol 70% for 1 minute and NaClO 3.5% for 30 seconds. This sterile stem was cut and then inoculated on the PDA. After 5 days, the fungi were transferred into the other PDA to give a single strain of fungus labeled with BTG-1.
Identification of fungus
The fungus with code BTG-1 was identified using analyses of the internal transcribed spacer (ITS) region of the ribosomal DNA. The fungus was cultivated on the PDB for 72 hours. The DNA was extracted from the mycelial of this fungus using nucleon PhytoPure and then amplified using a primer of ITS-4 and ITS-5 (Raja et al., 2017). The PCR (Polymerase Chain Reaction) product was purified with polyethylene glycol precipitation method (Hiraishi et al., 1995). The pure PCR product was sequenced with automated DNA sequencer. The sequencing data were trimmed and assembled with BioEdit program and then blasted at National Center for Biotechnology Information (NCBI).
Sequence ITS ribosomal DNA gene of fungus P. microspora
GAGGTCACCACAAAAAATTGGGGGTTTAGCGGCTGGGAGTTATAGCACCTAACAAAGCGAGAAAAAAATTACTACGCTCAGAGGATACTACAAATCCGCCGTTGTATTTCAGGAACTACAACTAATAAAAGAAGTAGATTCCCAACACTAAGCTAGGCTTAAGGGTTGAAATGACGCTCGAACAGGCATACCCACTAGAATACTAATGGGCGCAATGTGCGTTCAAAGATTCGATGATTCACTGAATTCTGCAATTCACATTACTTATCGCATTTCGCTGCGTTCTTCATCGATGCCAGAACCAAGAGATCCGTTGTTGAAAGTTTTGACTTATTAAAATAAGACGCTCAGATTACATAAAATAACAAGAGTTTAATGGTCCACCGGCAGCAGCTATAAGAAGACCTATAACTTCTGCCGAGGCAACAAAAGGTAAGTTCACATGGGTTGGGAGTTTAGAAAACTCTATAATG
Fermentation and isolation of secondary metabolites
Pestalotiopsis microspora HF 12440 was cultivated on PDB media and incubated at 27°C for 2 weeks. The mycelial and the filtrate were separated with Buchner funnel. The 10 l of the filtrate was extracted three times with ethyl acetate to give 2.2 g of ethyl acetate extract. The crude extract (2.2 g) was fractionated using vacuum liquid chromatography with the gradient polarity solvent system, i.e., dichloromethane, dichloromethane-acetone, acetone, and methanol to afford six fractions, FA, FB, FC, FD, FE, and FF.
A 210 mg of FB was subjected to radial chromatography using silica gel 60 PF254 eluted with hexane and acetone (8:2) to obtain compound 1 (11 mg) and compound 2 (20 mg). FC (314 mg) was fractionated by silica gel 60 PF254 eluted with hexane and ethyl acetate (from 6:4 to 4:6) to give 13 subfractions (FC.1–FC.13). Subfractions FC.11–FC.12 were purified by column chromatography using chloroform and acetone (from 9.5:0.5 to 6:4) to yield compound 3 (13 mg). A 211 mg of FD was submitted to column chromatography eluted with hexane, acetone, and ethyl acetate (7:2:1) to provide 23 subfractions (FD.1–FD.23). FD.9–FD.15 (59 mg) were combined and further separated by column chromatography using silica gel Kieselgel 60 eluted with chloroform and ethyl acetate (from 8:2 to 4:6) to afford compound 4 (6 mg).
Identification of secondary metabolites
The structures of compounds (1–4) were determined based on the analysis of spectroscopic data, including NMR (1D, 2D) and MS. NMR spectroscopic data were recorded at 500 MHz for 1H and 125 MHz for 13C with deuterated solvent peaks as reference standards and TMS as the internal standard. Each compound 1 and 3 was diluted with CDCl3 and each compound 2 and 4 was diluted with (CD3)2CO. The concentration of all compounds was 1.00 mg/ml. FID (Free Induction Decay) data were processed using MestreNova program.
(+)-Acetylpestalotin (1)
Colorless oil; +81.90 (c.0.0038, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.13 (1H, d, J = 1 Hz, H-3), 5.03 (1H, ddd, J = 4.0, 7.0, and 10.5 Hz, H-7), 4.45 (1H, dt, J = 3.5 and 12.5 Hz, H-6), 3.73 (3H, s, H-1′), 2.58 (1H, ddd, J = 2.0, 12.5, and 17.0 Hz, H-5a), 2.24 (1H, dd, J = 4.0 and 17.0 Hz, H-5b), 2.09 (3H, s, H-3′), 1.73 (2H, m, H-8), 1.30 (4H, m, H-9 and H-10), 0.89 (3H, t, J = 6.5 Hz, H-11); 13C NMR (125 MHz, CDCl3) (ppm): δ 172.7 (C-4), 170.7 (C-2′), 166.5 (C-2), 90.4 (C-3), 75.5 (C-6), 73.1 (C-7), 56.3 (C-1′), 29.7 (C-8), 29.4 (C-5), 27.6 (C-9), 22.6 (C-10), 21.0 (C-3′), 14.0 (C-11); MS m/z 257.29 [M + H]+ (calcd for C13H20O5, 257.29).
(−)-Pestalotin (2)
Yellow oil; -108.40 (c.0.0031, CHCl3); 1H NMR [500 MHz, (CD3)2CO]: δ 5.09 (1H, d, J = 1.5 Hz, H-3), 4.30 (1H, dt, J = 4.0 and 13.0 Hz, H-6), 3.78 (3H, s, H-1′), 3.61 (1H, m, H-7), 2.78 (1H, ddd, J = 2.0, 13.0, and 17.5 Hz, H-5a), 2.27 (1H, dd, J = 3.5 and 17.0 Hz, H-5b), 1.57 (2H, m, H-8), 1.47 (2H, m, H-9), 1.35 (2H, m, H-10), 0.91 (3H, t, J = 7.0 Hz, H-11); 13C NMR [125 MHz, (CD3)2CO] (ppm): δ 174.2 (C-4), 166.7 (C-2), 90.5 (C-3), 79.1 (C-6), 72.2 (C-7), 56.6 (C-1′), 33.0 (C-8), 29.8 (C-5), 28.7 (C-9), 23.3 (C-10), 14.3 (C-11); MS m/z 215.02 [M + H]+ (calcd for C11H18O4, 215.12).
(6S,7S,8R)-Hidroxypestalotin (3)
Yellow oil; -83.20 (c.0.00075, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.12 (1H, d, J = 1.2 Hz, H-3), 4.50 (1H, dt, J= 3.9 and 12.8 Hz, H-6), 3.78 (1H, m, H-8), 3.75 (3H, s, H-1′), 3.48 (1H, br s, H-7), 2.88 (1H, ddd, J = 1.2, 13.0, and 17.0 Hz, H-5a), 2.31 (1H, dd, J = 3.7 and 17.2 Hz, H-5b), 1.60 (1H, m, H-9a), 1.50 (2H, m, H-9b dan H-10a), 1.39 (1H, m, H-10b), 0.93 (3H, t, J = 7.2 Hz, H-11); 13C NMR (125 MHz, CDCl3) (ppm): δ 173.6 (C-4), 166.9 (C-2), 89.9 (C-3), 78.1 (C-6), 74.0 (C-7), 71.0 (C-8), 56.3 (C-1′), 36.1 (C-9), 29.5 (C-5), 18.9 (C-10), 14.1 (C-11); MS m/z 231.10 [M + H]+ (calcd for C11H18O5, 231.12).
(+)-Pinoresinol (4)
Colorless oil; +45.80 (c.0.0043, CHCl3); 1H NMR (500 MHz, (CD3)2CO) (ppm): δ 6.95 (d, J = 1.7 Hz, H-2′ and H-2″), 6.79 (dd, J = 1.7 and 8.1 Hz, H-6′ and H-6″), 6.74 (d, J = 8.1 Hz, H-5′ and H-5″), 4.62 (d, J = 4.1 Hz, H-2 and H-6), 4.16 (dd, J = 6.9 and 8.9 Hz, H-4a and H-8a), 3.80 (s, H-7′ and H-7″), 3.76 (dd, J = 3.7 and 9 Hz, H-4b and H-8b), 3.04 (m, H-1 and H-5); 13C NMR [125 MHz, (CD3)2CO]: 148.3 (C-3′ and C-3″), 146.9 (C-4′ and C-4’’), 134.2 (C-1′ and C-1″), 119.6 (C-5′ and C-5″), 115.6 (C-6′ and C-6″), 110.6 (C-2′ and C-2″), 86.6 (C-2 and C-6), 72.2 (C-4 and C-8), 56.2 (C-7′ and C-7″), 55.2 (C-1 and C-5); MS m/z 356.91 [M − H]+ (calcd for C20H22O6, 357.14).
Murine leukemia P388 cell assay
The crude extract and all pure compounds (1–4) were evaluated for their cytotoxicity against murine leukemia P388 cells using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide] assay as previously reported (Sahidin et al., 2005; Yang et al., 2008). The cells were seeded in 96-well plates (cell density 3 × 104 cells/cm3). Each compound (1–4) was added in various concentrations and incubated for 48 hours, where the crude extract and compounds were dissolved in dimethyl sulfoxide (DMSO). After 48 hours of incubation, 10 μl MTT reagent was added to each sample and then incubated for 4 hours. The MTT-stop solution containing sodium dodecyl sulfate (SDS) was added and the incubation was continued for 24 hours. Optical density was read with a microplate reader at 550 nm. IC50 values were taken from the plotted graph of percentage of live cells compared with control. The control was made from MTT solution and DMSO (without cells and medium).
RESULTS AND DISCUSSION
Compound 1 (11 mg) was obtained as colorless oil. Its molecular formula was determined as C13H20O5 by EI-MS spectrum (m/z 257.29 [M + H]+, calcd for 257.29, implying four degrees of unsaturation. The 1H NMR spectrum (see Supplementary Data) showed a triplet methyl at δH 0.89 ppm (J = 6.5 Hz), four methylene signals at δH 1.30, 1.73, 2.24, and 2.58 ppm, a singlet methyl (δH 2.09 ppm), a methoxyl signal (δH 3.73 ppm), two oxygenated methine signals at δH 4.45 and 5.03 ppm, and a methine (δH 513 ppm, d, 1 Hz). The 13C NMR data showed that this compound consists of 13 carbons, including three methyls, four methylenes, three methines (two oxygenated), and three quaternary carbons. The structure of compound 1 was confirmed by HMBC spectrum. The methoxyl (δH 3.73 ppm, H-1′), methine at δH 5.13 ppm (H-3), and methylene (δH 2.24 and 2.58 ppm, H-5) correlated to quaternary carbon at δC 172.7 ppm (C-4). It afforded information on the presence of methoxyl at quaternary carbon, C-4. An oxygenated methine at δH 5.03 ppm (H-7) and singlet methyl (δH 2.09 ppm, H-3′) correlated to δC 170.7 ppm (C-2′) indicated the presence of acetyl at C-7. Based on the correlation (Fig. 2), the structure of compound 1 was suggested as 6-[1-(acetyloxy)pentyl]-5,6-dihydro-4-methoxy-2H-pyran-2-one. The stereocenters of C-6 and C-7 in compound 1 were determined by comparison of the optical rotation data (1, +81.90, c.0.0038, CHCl3; (+)-pestalotin, +92.50, c.1.30, MeOH; (−)-pestalotin, -94.760, c.1.00, MeOH) (Akay et al., 2014; Kumar et al., 2005; Mayer et al., 2002). On the basis of comparison of this data with the literature, the stereochemistry of compound 1 was assigned as C-6(R) and C-7(R). Compound (1) is a new compound.
A 20 mg of compound 2 was isolated as yellow oil. EI-MS showed ion peak at m/z 215.02 [M + H]+ indicating that compound 2 had a molecular formula C11H18O4. The spectrum of 1H and 13C NMR of compound 2 were similar with compound 1 (Table 1), except the oxymethine proton (H-7) at δH 3.61 ppm arised more shielding than that of compound 1 (δH 5.03 ppm). The change in chemical shift of 1H NMR revealed the absence of acetyl group at C-7 in compound 2. The methylene proton at position 5 and 8 correlated in HMBC spectrum to the two oxymethine carbons at δC 72.2 and 79.1 ppm (C-7 and C-6). An olefinic proton (δH 5.09 ppm, H-3) correlated to the two quaternary carbons (δC 166.7 and 174.2 ppm). The selected HMBC correlations of compound 2 were displayed in Figure 2. The spectroscopic data of compound 2 were similar to literature (Akay et al., 2014). Absolute configurations of C-6 and C-7 in compound 2 were assigned as S and S based on the comparing of the optical rotation and NMR data with the literature (2, -108.40, c.0.0031, CHCl3; (+)-pestalotin, + 92.50, c.1.30, MeOH; (−)-pestalotin, -94.760, c.1.00, MeOH) (Akay et al., 2014; Kumar et al., 2005; Mayer et al., 2002). Therefore, compound 2 was elucidated to be (6S)-6-[(1S)-1-hydroxypentyl]-4-methoxy-5,6-dihydro-2H-pyran-2-one and named (−)-pestalotin.
Compound 3 (13 mg) was acquired as the yellow oil. EI-MS showed mass ion peak at m/z 231.10 [M + H]+, establishing the molecular formula C11H18O5 with three degrees of unsaturation. 1H and 13C NMR spectrum of compound 3 were conformable with compound 2 (Table 1), except the proton (δH 3.78 ppm) and carbon (δC 71.0 ppm) at position 8 appeared more deshielding than that compound 2 (δH 1.57 ppm and δC 33.0 ppm) because of the presence of hydroxyl at this position. The position of hydroxyl at C-8 was also confirmed by the HMBC spectrum (Fig. 2). Methylene proton at δH 1.50 and 1.60 ppm (H-9) correlated to the two oxymethine carbon (δC 71.0 and 74.0 ppm, C-8 and C-7). The configurations of chiral carbon at the position C-6, C-7, and C-8 in compound 3 were determined as (S), (S), and (R) based on the comparing of the optical rotation and NMR data with the literature (3, − 83.20, c.0.00075, CHCl3); (6S,7S,8R)-LL-P880β, -59.80, c.0.96, MeOH) (Kirihata et al., 1990). The structure of compound 3 was displayed in Figure 1.
Compound 4 (6 mg) was obtained as colorless oil. The spectrum of EI-MS showed ion peak at m/z 356.91 [M − H]+. The molecular formula of compound 4 was decided as C20H22O6. There are 10 signals in the spectrum of 13C NMR representing of 20 carbons, consist of four methines (two oxygenated), two methoxyls, two oxymethylenes, and 12 carbons from two benzenes (six quaternary carbons). 1H NMR showed two methines at δH 3.04 ppm (2H, q), two oxymethilene protons (δH 3.76 and 4.16 ppm, 4H), two methoxyls at δH 3.80 ppm (6H, s), two oxygenated methine protons (δH 4.62 ppm, 2H), and three aromatic proton signals with ABX system at δH 6.74 ppm (2H, d, 8.1 Hz), 6.79 ppm (2H, dd, 1.7 and 8.1 Hz), and 6.95 ppm (2H, d, 1.7 Hz). In the HMBC spectrum, the methoxyl (δH 3.80 ppm, H-7′ and H-7″) correlated to the quaternary carbon at δC 148.3 ppm (C-3′ and C-3″), indicating the presence of methoxyl at C-3′ and C-3″. The correlation of methine (δH 3.04 ppm, H-1 and H-5) and oxygenated methine proton (δH 4.62 ppm, H-2 and H-6) with a quaternary carbon at δH 134.2 ppm (C-1′ and C-1″) implied the existence of benzyl at the C-2 and C-6 of the furan ring. The selected HMBC correlations of compound (4) were shown in Figure 3. The optical rotation and NMR data were used for determining the stereochemistry of compound 4 (4, +45.80, c.0.0043, CHCl3); (+)-pinoresinol, + 64.00, c.1.0, CHCl3; (−)-pinoresinol, -34.7 0, c.0.91, CH3OH) (Brenes et al., 2000; Fonseca et al., 1979; Lin-gen et al., 1982). The result of comparing this data with all literatures showed compound 4 was (+)-pinoresinol. Interestingly, this is the first reported of (+)-pinoresinol (4) isolated from endophytic fungi.
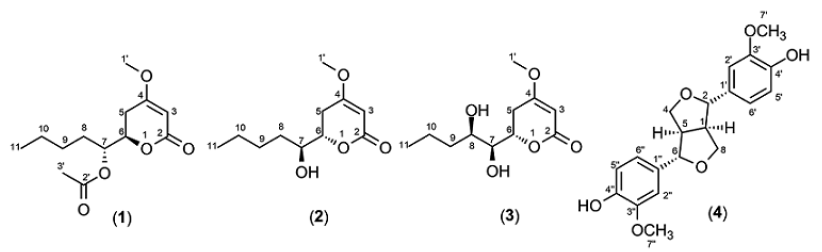 | Figure 1. Structures of compounds (1–4) from ethyl acetate extract of P. microspora HF 12440. [Click here to view] |
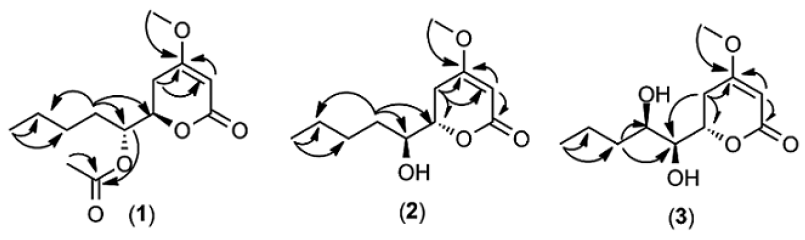 | Figure 2. Selected HMBC (H→C) correlation of compounds 1, 2, and 3. [Click here to view] |
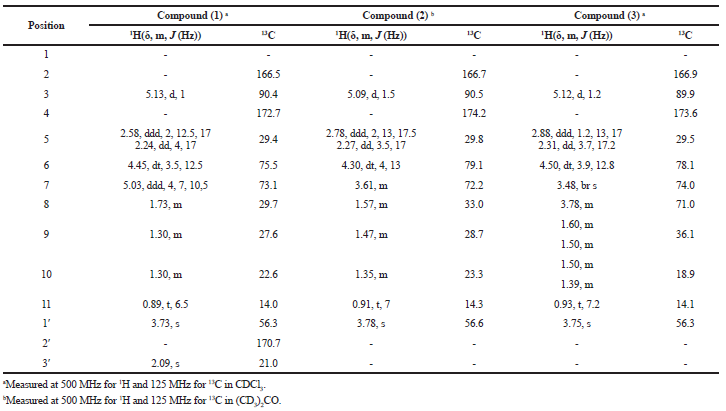 | Table 1. 1H and 13C data of compounds 1, 2, and 3. [Click here to view] |
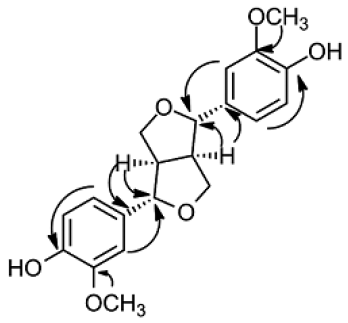 | Figure 3. Selected HMBC correlation of compound 4. [Click here to view] |
The crude extract and all compounds (1–4) were assayed for their cytotoxicity against murine leukemia P388 cells followed the MTT method. Compound (3), (4), and the crude extract displayed significant cytotoxicity with IC50 3.34; 3.62; 18.97 μg/ml, respectively. Meanwhile, compound (1) and (2) showed moderate cytotoxicity with IC50 values of 5.60 and 7.1 μg/ml, respectively. The presence of hydroxyl group at C-8 in compound 3 is an important factor for cytotoxic value. Interestingly, compound 1 is much less active because of the absence of a hydroxyl group and the stereochemistry of this compound is different.
CONCLUSION
A new lactone (1), (+)-acetylpestalotin, together with three known compounds, (−)-pestalotin (2), (6S,7S,8R)-hydroxypestalotin (3), and (+)-pinoresinol (4) were isolated from the liquid media of P. microspora HF 12440, an endophytic fungus from the stem of A. heterophyllus. Crude extract and two compounds (3–4) were active against murine leukemia P388 cells with IC50 values of 18.97, 3.34, and 3.62 μg/ml, respectively.
ACKNOWLEDGMENTS
PMDSU Scholarship (Ministry of Research, Technology and Higher Education of Indonesia) for financial support (grant number: 1511/E4.4/2015); Indonesian Institute of Science for the identification of endophytic fungus; Prof. Dr. Yana Maolana Syah and Dr. Elvira Hermawati for their aid in obtaining the NMR and mass spectra at the Bandung Institute of Technology.
CONFLICT OF INTEREST
The authors declare that no conflict of interest is associated with this work.
REFERENCES
Achmad SA, Hakim EH, Juliawaty LD, Makmur L, Suyatno. A new prenylated flavone from Artocarpus champeden. J Nat Prod, 1996; 59:878–9.
Akay S, Ekiz G, Kocabas F, Cabas EH, Korkmaz KS, Bedir E. A new 5,6-dihydro-2-pyrone deivative from Phmopsis amygdali, an endophytic fungus isolated from hazelnut (Corylus avellana). Phytochem Lett, 2014; 7:93–6.
Brenes M, Hidalgo FJ, Garcia A, Rios JJ, Garcia P, Zamora R, Garrido A. Pinoresinol and 1-acetxypinoresinol, two new phenolic compound in olive oil. J Am Oil Chem Soc, 2000; 77:715–20.
Fonseca SF, Nielsen LT, Ruveda EA. Lignans of Araucaria angustifolia and 13C NMR analysis of some phenyltetralin lignans. Phytochemistry, 1979; 18:1703–8.
Gao XX, Zhou H, Xu DY, Yu CH, Chen YQ, Qu LH. High diversity of endophytic fungi from the pharmaceutical plant, Heterosmilax japonica Kunth revealed by cultivation-independent approach. FEMS Microbiol Lett, 2005; 249:255–66.
Guo BH, Wang YC, Sun X, Tang K. Bioactive natural products from endophytes: a review. Appl Biochem and Micro+, 2008; 44:153–8.
Hakim EH, Achmad SA, Juliawaty LD, Makmur L, Syah YM, Aimi N, Kitajima M, Takayama H, Ghisalberty EL. Prenylated flavonoids and related compounds of the Indonesian Artocarpus (Moraceae). J Nat Med, 2006; 60:161–84.
Hakim EH, Asnizar, Yurnawilis, Aimi N, Kitajima M, Takayama H. Artoindonesianin P, a new prenylated flavone with cytotoxicity from Artocarpus lanceifolius. Fitoterapia, 2002; 73:668–73.
Hakim EH, Fahriyati, Kau MS, Achmad SA, Makmur L, Ghisalberti EL, Nomura T. Artoindonesianins A and B, two new prenylated flavones from the root bark of Artocarpus champeden. J Nat Prod, 1999; 62:613–5.
Hiraishi A, Kamagata Y, Nakamura N. Polymerase chain reaction amplification and restriction fragment length polymorphism analysis of 16S rRNA gene from methanogens. J Ferment Bioeng, 1995; 79:523–9.
Jagtap. Artocarpus: a review of its traditional uses, phytochemistry and pharmacology. J Ethnopharmacol, 2010; 129:142–166.
Jia M, Chen L, Xin HL, Zheng CJ, Rahman K, Han T, Qin LP. A friendly relationship between endophytic fungi and medicinal plants: a systematic review. Front Microbiol, 2016; 7:1–14.
Kirihata M, Ohta K, Ichimoto I, Ueda H. Total synthesis of (6S,1’S,2’R)-6-(1’,2’-dihydroxypentyl)-4-methoxy-5,6-dihidroxypyrane-2-one (LL-P880β) and its C6 epimer, a fungal metabolite from Penicillium sp. Agr Biol Chem, 1990; 54:2401–5.
Kogel KH, Franken P, Huckelhoven R. Endophyte or parasite what decides? Curr Opin Plant Biol, 2006; 9:358–63.
Kuang C, Jing SX, Liu Y, Luo SH, Li SH. Drimane sesquiterpenoids and isochromone derivative from the endophytic fungus Pestalotiopsis sp. M-23. Nat Prod Bioprospec, 2016; 6:155–60.
Kumar AS, Bhaket P, Rao BV. Stereoselective synthesis of (-)-pestalotin. Arkivoc, 2005; 3:74–82.
Kyekyeku JO, Kusari S, Adosraku RK, Bullach A, Golz C, Strohman C, Spiteller M. Antibacterial secondary metabolites from an endophytic fungus, Fusarium solani JK10. Fitoterapia, 2017; 119:108–14.
Lin-gen Z, Seligmann O, Jurcic K, Wagner H. Constituents of Daphne tangutica. Planta Med, 1982; 45:172–6.
Mayer SF, Steinreiber A, Goriup M, Saf R, Faber K. Chemoenzymatic asymmetric total syntheses of a constituent of Jamaican rum and of (+)-pestalotin using an enantioconvergent enzyme-triggered cascade reaction. Tetrahedron Asymmetr, 2002; 13:523–8.
Müller JL. Plants and endophytes: equal partners in secondary metabolite production ?. Biotechnol Lett, 2015; 15:1814.
Musthapa I, Hakim EH, Juliawaty LD, Syah YM, Achmad SA. Prenylated flavone from Indonesian Artocarpus and their antimalarial properties. Med Plants, 2010; 2:145–8.
Mustapha I, JuliawatyLD, Syah YM, Hakim EH, Latif J, Ghisalberti EL. An oxepinoflavone from Artocarpus elasticus with cytotoxic activity against P-388 cells. Arch Pharm Res, 2009; 32:191–4.
Raja HA, Miller AN, Pearce CJ, Oberlies NH. Fungal identification using molecular toolls: a primer for natural products research community. J Nat Prod, 2017; 80:756–70.
Sahidin, Hakim EH, Juliawaty LD, Syah YM, Din LL, Ghisalberti EL, Said IM, Achmad SA. Cytotoxic properties of oligostilbenoids from the tree barks of Hopea dryobalanoides. Z. Naturforsch, 2005; 60:723–7.
Septama AW, Panichayupakaranant P. Artocarpin isolated from Artocarpus heterophyllus heartwoods alters membrane permeability of Streptococcus mutans. J Appl Pharm Sci, 2018; 8:059–64.
Stone JK, Polishook JD, White JRJ. Biodiversity of fungi: inventory and monitoring methods. Burlington, Elsevier, Boston, USA, 2004; 241–70.
Syah YM, Achmad SA, Ghisalberti EL, Hakim EH, Makmur L, Mujahidin D. Artoindonesianins G-I, three new isoprenylated flavones from Artocarpus lanceifolius. Fitoterapia, 2001; 72:765–73.
Syah YM, Achmad SA, Ghisalberti EL, Hakim EH, Mujahidin D. Two new cytotoxic isoprenylated flavones, artoindonesianins U and V, from the heartwood of Artocarpus champeden Spreng. (Moraceae). Fitoterapia, 2004; 75:134–40.
Wei B, Yang ZD, Chen X, Zhou SY, Yu HT, Sun JY, Yao XJ, Wang Y, Xue HY. Colletotrilactam A-D, novel lactams from Colletotrichum gloeospoirioides GT-7, a fungal endophyte of Uncaria rhyncophylla. Fitoterapia, 2016; 113:158–63.
Xia X, Kim S, Liu C, Shim SH. Secondary metabolites produced by an endophytic fungus Pestalotiopsis sydowiana and their 20S proteasome inhibitory Activities. Molecules, 2016; 21:944.
Xiao J, Lin L, Hu J, Jiao F, Duan D, Zhang Q, Tang H, Gao J, Wang L, Wang X. Highly oxygenated caryophyllene-type and drimane-type sesquiterpenes from Pestalotiopsis adusta, an endophytic fungus of Sinopodophyllum hexandrum. RSC Adv, 2017; 7:29071–9.
Xu J, Ebada SS, Proksch P. Pestalotiopsis a highly creativegenus: chemistry and bioacivity of secondary metabolites. Fungal Divers, 2010; 44:15–31.
Yang RL, Yan ZH, Lu Y. Cytotoxic phenylpropanoids from carrot. J Agr Food Chem, 2008; 56:3024–7.
Zhang HW, Song YC, Tan RX. Biology and chemistry of endophytes. Nat Prod Rep, 2006; 23:753–71.