
INTRODUCTION
Dipeptidyl peptidase-IV (DPP IV) is a protease responsible for in vivo inactivation of several endogenous peptides including glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1). GLP-1 and GIP are incretins released from the gut in response to intraluminal glucose and play an important role in glucose homeostasis (Campbell and Drucker, 2013; Drucker, 2003). Furthermore, they inhibit apoptosis of β-cells and enhance their regeneration (Brubaker and Drucker, 2004; Yabe and Seino, 2011). However, the effects of these incretins in diabetic patients are significantly reduced and suppression of DPP IV has been shown to improve glycemic control by enhancing the insulinotropic effects of incretins (Karagiannis et al., 2014; Kazafeos, 2011). Therefore, the search for DPP IV inhibitors, as antidiabetic agents, is an active area of research (Almasri et al., 2013; Shu et al., 2014).
Both reversible and irreversible DPP IV inhibitors with in vivo efficacy were discovered. The reversible substrate analogs inhibitors take benefit of the high preference of the S1 hydrophobic site of DPP IV for proline binding. These inhibitors are generally amide derivatives of pyrrolidine or thiazolidine. Examples include valine-pyrrolidide and isoleucine-thiazolidide (Fig. 1) (Peters, 2011). The irreversible inhibitors contain, in addition to the five-membered ring, an electrophilic group at position 2 and are even more potent. Nitrile group is the most common electrophilic group used and is capable of forming an enzyme-imidate adduct with Ser630 within the active site and is present in saxagliptin and vildagliptin (Fig. 1) (Kushwaha et al., 2014).
Sitagliptin is an example on reversible non-peptide heterocyclic inhibitors (Fig. 1) (Kim et al., 2005; Liu et al., 2012). It does not contain a pyrrolidine ring to bind within the S1 pocket. As an alternative, sitagliptin has a pyrazole-piperazine fused ring system and binds to the enzyme so that the 2,4,5-trifluorophenyl group binds within the S1 hydrophobic site and the amide group is in the opposite direction to that of substrate analog inhibitors (Kim et al., 2005). Xanthine, aminomethylpyrimidine, and isoquinoline DPP IV inhibitors are another examples on reversible inhibitors in which the amide bond is not necessary for activity (Kushwaha et al., 2014). Several antidiabetic drugs have recently been explored for their effect on DPP IV. Metformin has been reported to inhibit DPP IV (Kaganda et al., 2015; Lindsay et al., 2005) with IC50 ranging from 29 to 98 µM. Thiazolidinediones derivatives, e.g., rosiglitazone and troglitazone; meglitinides, e.g., nateglinide; sulfonylureas, e.g., glybenclamide and tolbutamide have also been reported to inhibit DPP IV (Duffy et al., 2007). However, to date no work was done to thoroughly investigate the effect of other non-diabetic drugs on DPP IV activity.
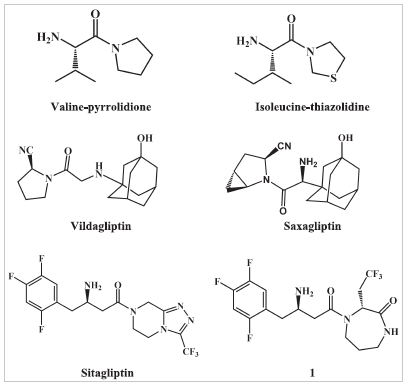 | Figure 1. Structures of different classes of DPP IV inhibitors. [Click here to view] |
Herein, structure-based virtual screening approach was employed to screen in house built drug database for hits capable virtually to fit within the DPP IV binding pocket in order to identify drugs with potential anti-DPP IV activities. High throughput FRED docking software was used as a database screening tool.
METHODOLOGY
Molecular docking
The 3D coordinates of DPP IV were obtained from the Protein Data Bank (PDB code: 2G63, resolution 2.0Å) (Pei et al., 2007). Hydrogen atoms were added to the protein structure and the co-crystallized ligand was extracted using Discovery studio Visualizer. The protein structure was exploited in subsequent docking experiments in the presence of explicit water and without energy minimization. High throughput docking/scoring technique using FRED docking software (FRED, 2006) was employed to screen in house built drug database (1,490 drugs) for hits capable virtually to fit within the DPP IV binding pocket of co-crystallized complex. Although further details about FRED docking are described elsewhere (Almasri et al., 2008; Bustanji et al., 2009), a brief description is provided below. FRED docking program requires a target protein structure, a box defining the active site of the target, a multi-conformer database of the ligand(s) intended for docking and a number of optional parameters as input. The conformational space of the database drugs was explored employing OMEGA program (OMEGA, 2013). The protein structure and ligands conformers are treated as rigid structures during the docking experiment. Upon completion of docking calculation, poses are scored and ranked. The top-scored pose was chosen using the ScreenScore function. The software parameters that best retrieved the co-crystallized pose of DPP IV complex were employed in the docking experiment. The docking experiment was validated by extracting the crystallographic bound cyanopyrrolidine inhibitor and redocking it to the binding site of DPP IV. This validation resulted in very close model to the crystallographic structure.
In vitro DPP IV enzyme inhibition assay
The assay was conducted using DPP IV Drug Discovery Kit (Biomol, Germany) in which DPP IV inhibition activity was quantified by measuring the release of para-nitroaniline (pNA) from chromogenic substrate (H-Gly-Pro-para-nitroaniline) by DPP IV as previously described (Almasri et al., 2008).
Briefly, fexofenadine was dissolved in DMSO and diluted with Tris buffer solution (pH 7.5). The Recombinant enzyme was diluted in the assay buffer solution to obtain a final concentration of 17.34 µUml−1. Afterward, appropriate volumes of fexofenadine stock solution were added to a 15 µl aliquot of the enzymatic solution into microplate wells and the volume completed to 50 µl with buffer. The obtained mixtures were incubated for 10 minutes at 37oC. Finally, 50 µl of 0.20 mM substrate solution was added to each well. The absorbance of the plate was read at 405 nm using microplate reader (BioTek) and the rate of absorbance reduction was monitored over 10 minutes and compared with a negative control. P32/98, a standard DPP IV inhibitor, was used as positive control. The DMSO concentration was kept less than 1.0% in all runs.
The absorbance versus time was plotted and the slope and percent inhibition were calculated using the following formula:
Percent inhibition = (1 − slope of the compound/slope of the negative inhibitor)*100%.
Oral glucose tolerance test (OGTT) in mice
The animal experimental protocol comply with the guide of laboratory animals use (National Research Council, 2011). Male Balb/c mice 25–30 g obtained from Jordan University animal house were used in the OGTT. Both control and treatment groups were matched for body weight in all experiments. A dose of 34 mg/kg of fexofenadine was given intraperitoneally to Balb/c mice 30 minutes prior to oral glucose load evaluation. Mice were fasted for 6 hours at the end of the light/dark cycle before the test. Glucose solution was administered orally at a dose of 2 g/kg of body weight, subsequently, the blood samples were collected at 0, 15, 30, 60, and 120 minutes and analyzed for glucose level using glucometer (Arkray, Inc., Japan).
RESULTS AND DISCUSSION
The identification of lead compounds having some activity against a biological target and progressive optimization of the potency and physicochemical properties of these compounds are key steps in the early-stage drug discovery. Virtual screening (VS) is one of the most widely used powerful techniques in lead discovery. The fundamental goal of VS is to reduce the huge virtual chemical space of library of molecules to a manageable number of compounds to screen against a specific target protein (Shoichet, 2004). Two common strategies are usually adopted in database searching: structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS). In SBVS, we make use of the 3D structural information of the target protein and computational techniques in virtual database screening and to study the essential molecular interactions involved in ligand-protein binding, and thus explain experimental results at the molecular level. Moreover, the use of VS in drug discovery has additional advantages of lowering the cost and speed up the process of delivering new drugs into the market.
In the current work, molecular docking was employed in virtually screening a drug database. Interestingly, one of the highest ranking hits was fexofenadine, a second generation antihistamine drug (Fig. 2). Although fexofenadine structure differs from known DPP IV inhibitors, it binds to DPP IV (Fig. 4A) in a comparable way to known DPP IV inhibitors and interacts with the same essential key amino acids (Almasri et al., 2008). In the present work, docking simulations revealed that fexofenadine binds to the active site of DPP IV with one of the two phenyl moieties laid within the hydrophobic S1 pocket formed by Tyr547, Tyr631, Val656, Trp659, Tyr662, Tyr666, Val711, and His740. A major contribution to binding by this phenyl moiety is achieved by edge-to-face π–π interaction with Tyr666 and face-to-face π–π interaction with Tyr662. These interactions are comparable to the co-crystallized ligand cyanopyrrolidine ring interactions within the S1 pocket (Fig. 4B) (Pei et al., 2007). The entry of DPP IV substrate within the S1 pocket will be hindered because of the binding of phenyl ring within the same cavity. Furthermore, the key amino acid Tyr547 was engaged in aromatic stacking interaction with terminal para-substituted phenyl ring in a similar manner to the co-crystallized ligand aromatic ring (Fig. 4). Moreover, the pyrrolidine nitrogen of the co-crystallized structure is strongly attracted via a hydrogen-bond-reinforced ionic interaction with Glu205 (2.0 Å) and Glu206 (3.8 Å) and similarly the positively ionizable nitrogen of the piperidine ring is within the electrostatic interaction range with the key amino acid Glu206 (6.0 Å). The hydroxyl group attached to the quaternary carbon is hydrogen bonded to the amino group of Asn710 (2.3 Å), which are part of the P2 region, and to Tyr662 (2.5 Å) similar to carbonyl group interaction in the co-crystallized ligand. Additionally, the secondary hydroxyl group is hydrogen-bonded to Tyr547 (3.2 Å) comparable to the cyano group in the co-crystallized structure. The hydroxyl group of Tyr547 was found to play a role in the stabilization of the negatively charged oxyanion of the formed intermediate via hydrogen bonding (Thoma et al., 2003). Finally, the hydrocarbon butyl chain of fexofenadine is involved in hydrophobic (van der Waals) interactions with Phe357. The obtained docking results have encouraged us to evaluate the anti-DPP IV activity of fexofenadine in vitro. The IC50 of the drug against DPP IV was 4.6 (± 1.0) μM (Fig. 3).
To substantiate the anti-DPP IV activity of fexofenadine, it was decided to test its ability to decrease blood glucose level in Balb/c mice using OGTT. Intraperitoneal administration of fexofenadine (34 mg/kg) inhibits glucose excursion, as shown in Figure 5A. Furthermore, it illustrated significant reduction in the AUC of plasma glucose/time curve at three dosing levels (Fig. 5B).
The discovered binding mode of fexofenadine put forward several ways for more improvements in its activity. Replacing hydroxyl group attached to quaternary carbon by NH2 group could enhance the drug binding via forming a strong hydrogen-bond-reinforced ionic interaction with both Glu205 and Glu206 and hydrogen bond with Tyr662. Moreover, rigidifying or shortening the butyl linker is expected to enhance the activity. It has been shown that the free energy of binding is reduced by approximately 0.7 kcal/mol for each freely rotating bond in a ligand (Andrews et al., 1984). Several success stories are reported in the literature were rigidification of a flexible ligand leads to significant increase in affinity. Moreover, removing the second aromatic ring, which is attached to the quaternary carbon and does not make interactions within the active site, would decrease the molecular weight, and thus increase ligand efficiency (Reynolds et al., 2008).
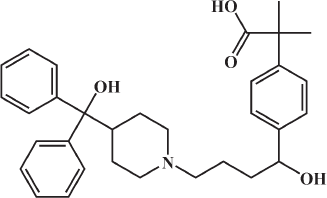 | Figure 2. The chemical structure of fexofenadine. [Click here to view] |
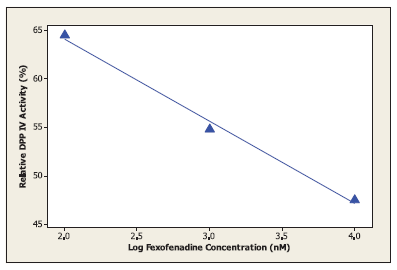 | Figure 3. The effect of variable fexofenadine concentrations on the activity of dipeptidyl peptidase IV (DPP IV). [Click here to view] |
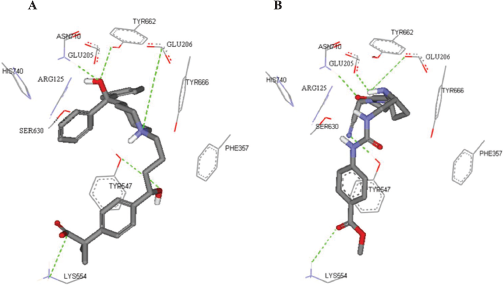 | Figure 4. The highest ranking FRED predicted pose for fexofenadine (A) compared to the co-crystallized cyanopyrrolidine ligand (B) within the catalytic site of DPP IV (PDB code: 2G63, resolution 2.0). For clarity, only polar hydrogen atoms are shown. Hydrogen bonds are shown as dotted green lines. Catalytic site residues are represented as sticks with the atoms in standard color. [Click here to view] |
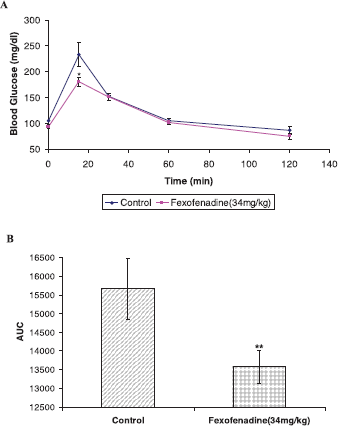 | Figure 5. Oral glucose tolerance test of fexofenadine using male Balb/c mice. A dose of 34 mg/kg of fexofenadine was administered intraperitoneally to Balb/c mice, and glucose (2 g/kg, p.o.) was given 30 minutes latter (0 minutes). Plasma glucose concentration was measured at the indicated time (A). In A, (♦) Control (DMSO), (â– ) fexofenadine 34 mg/kg. Data are represented as mean ± standard error of the mean. (B). AUC of glucose concentration curve (A) was calculated and displayed. *p value < 0.05 vs. control, **p value < 0.01 vs. control. [Click here to view] |
CONCLUSION
Docking simulations adopted in this work revealed fexofenadine as potential lead inhibitor for the anti-diabetic target DPP IV in vitro. Furthermore, the anti-diabetic effect of fexofenadine was validated by the in vivo OGTT. Moreover, the obtained results in this study could be helpful in the discovery of new fexofenadine-like DPP IV inhibitors.
ETHICAL APPROVAL
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
Almasri IM, Taha MO, Mohammad MK. New leads for DPP IV inhibition: structure-based pharmacophore mapping and virtual screening study. Arch Pharm Res, 2013; 36(11):1326–37.
Almasri IM, Mohammad M, Taha MO. Discovery of new DPP IV inhibitors via pharmacophore modeling and QSAR analysis followed by in-silico screening. ChemMedChem, 2008; 3:1763–79.
Andrews PR, Craik DJ, Martin JL. Functional group contributions to drug-receptor interactions. J Med Chem, 1984; 27:1648–57.
Brubaker PL, Drucker DJ. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology, 2004; 145:2653–9.
Bustanji Y, Taha MO, Almasri IM, Al-Ghussein M, Mohammad M, AlKhatib H. Inhibition of glycogen synthase kinase by curcumin: investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J Enzyme Inhib Med Chem, 2009; 24:771–8.
Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab, 2013; 4:819–37.
Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care, 2003; 26:2929–40.
Duffy NA, Green BD, Irwin N, Gault VA, McKillop AM, O’Harte FPM, Flatt PR. Effects of antidiabetic drugs on dipeptidyl peptidase IV activity: nateglinide is an inhibitor of DPP IV and augments the antidiabetic activity of glucagon-like peptide-1. Eur J Pharmacol, 2007; 568:278–86.
FRED 2.1.5. OpenEye scientific software, Santa Fe, NM, 2006. Available via http://www.eyesopen.com (Accessed August 2009).
Kaganda O, Singh C, Sachdeva K. Recent advancement in treatment of type-II diabetes mellitus: a review. Int J Pharmacy Pract Pharm Sci, 2015; 2:4–14.
Karagiannis T, Boura P, Tsapas A. Safety of dipeptidyl peptidase 4 inhibitors: a perspective review. Ther Adv Drug Saf, 2014; 5:138–46.
Kazafeos K. Incretin effect: GLP-1, GIP, DPP4. Diabetes Res Clin Pract, 2011; 93:S32–36.
Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, Marsilio F, McCann ME, Patel RA, Petrov A, Scapin G, Patel SB, Roy RS, Wu JK, Wyvratt MJ, Zhang BB, Zhu L, Thornberry NA, Weber AE. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem, 2005; 48:141–51.
Kushwaha RN, Haq W, Katti SB. Discovery of 17 gliptins in 17-years of research for the treatment of type 2 diabetes: a synthetic overview. Chem Biol Interface, 2014; 4:137–62.
Lindsay JR, Duffy NA, McKillop AM, Ardill J, O’Harte FPM, Flatt PR, Bell PM. Inhibition of dipeptidyl peptidase IV activity by oral metformin in Type 2 diabetes. Diabetic Med, 2005; 22:654–7.
Liu Y, Hu Y, Liu T. Recent advances in non-peptidomimetic dipeptidyl peptidase 4 inhibitors: medicinal chemistry and preclinical aspects. Curr Med Chem, 2012; 19:3982–99.
National Research Council. Guide for the care and use of laboratory animals. 8th edition, National Academies Press, Washington, DC, 2011.
OMEGA 2.5.1.4. OpenEye scientific software, Santa Fe, NM, 2013. Available via http://www.eyesopen.com (Accessed August 2009).
Pei Z, Li X, Geldern TW, Longenecker K, Pireh D, Stewart KD, Backes BJ, Lai C, Lubben TH, Ballaron SJ, Beno DW, Kempf-Grote AJ, Sham HL, Trevillyan JM. Discovery and structure-activity relationships of piperidinone- and piperidine-constrained phenethylamines as novel, potent, and selective dipeptidyl peptidase IV inhibitors. J Med Chem, 2007; 50:1983–7.
Peters JU. 11 years of cyanopyrrolidines as DPP-IV inhibitors. Curr Top Med Chem, 2007; 7:579–95.
Reynolds CH, Tounge BA, Bembenek SD. Ligand binding efficiency: trends, physical basis, and implications. J Med Chem, 2008; 51:2432–8.
Shoichet BK. Virtual screening of chemical libraries. Nature, 2004; 432:862–5.
Shu C, Ge H, Song M, Chen JH, Zhou H, Qi Q, Wang F, Ma X, Yang X, Zhang G, Ding Y, Zhou D, Peng P, Shih CK, Xu J, Wu F. Discovery of imigliptin, a novel selective DPP-4 inhibitor for the treatment of type 2 diabetes. ACS Med Chem Lett, 2014; 5(8):921–6.
Thoma R, Löffler B, Stihle M, Huber W, Ruf A, Hennig M. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure, 2003; 11:947–59.
Yabe D, Seino Y. Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and β cell preservation. Prog Biophys Mol Biol, 2011; 107:248–56.