
INTRODUCTION
Inflammation is an important process of a body’s defense system in response to damage to the tissues and cells by pathogens, noxious stimuli, such as chemicals or physical injury, which acts to repair or removal of the damaged tissue or neutralizes the harmful effects (Ferrero et al., 2006; Maslinska et al., 1998). Inflammatory response is characterized by endothelial permeability, platelet aggregation to the vessels in the vicinity of the injured site, migration of several cell types, the growth of new tissues and blood vessels. Inflammation is mediated by secretion of pro-inflammatory cytokines like bradykinins, serotonin, histamines, prostaglandins, and nitric oxide. These pro-inflammatory mediators contribute to the clinical picture of redness, pain, swelling, and heat. The expression of many proteins, including pro-inflammatory cytokines, chemokines, and enzymes of the arachidonic acid cascade is regulated by the transcriptional factor nuclear factor kappa B (NF-κB) (Karin et al., 2005; Pahl et al., 1999). Hence, NF-κB is considered as the primary regulator of the inflammatory process.
NF-κB is a nuclear factor of kappa-light-chain-enhancer of activated B cells which exists as a heterodimer and involved in the regulation of cell signaling responses and plays a vital role in the regulation of cellular processes involved in immune response, differentiation, cell proliferation, and apoptosis (Baldwin et al., 1996). The family of NF-κB transcriptional factor is intermediately involved in the regulation and expression of numerous genes in the setting of the inflammatory response. The members of the NF-κB family include p65 (Rel-A), Rel-B, c-Rel, P50/P105 (NF-κB1), and P52/P65 (NF-κB2). NF-κB1 and NF-κB2 are synthesized as a large precursor P105/P100, which is post-translationally modified to P50 and P52 (Ghosh et al., 1998). All the members of the NF-κB family have N-terminal Rel homology domain that allows DNA binding and dimerization and nuclear localization in common. Many in vitro and in vivo studies have reported the contribution of components in the NF-κB signaling pathway to the pathogenesis of various ailments. NF-κB plays an important role in inflammation and cell proliferation, angiogenesis, and pannus formation.
NF-κB is activated by different inflammatory stimuli, such as bacterial infection, lipopolysaccharides, injury, UV radiation, reactive oxygen species (ROS) and inflammatory cytokines (tumor necrosis factor-α; TNF-α), and interleukin-6 (IL-6) through phosphorylation by IKappa B kinase and proteasome degradation of the IκB complex. The activated NF-κB is translocated into the nucleus which activates the transcription of various genes to synthesize different proinflammatory mediators, such as cytokines, chemokines, adhesion molecules, phospholipids, lipoxygenases, cyclooxygenases, inducible nitric oxide synthase (iNOS), and myeloperoxidases. Abnormal activation of the NF-κB pathway is involved in degenerative chronic inflammatory diseases, such as asthma, rheumatoid arthritis, atherosclerosis, Alzheimer’s, and inflammatory bowel disease (Collins et al., 2001; Tak et al., 2001). NF-κB has an evolutionary conserved domain that plays an important role in many biological processes in addition to inflammation through signaling systems.
Diabetes mellitus is the most common endocrine disorder in humans. Besides β-cell failure, the major pathophysiological event contributing to type-2 diabetes (T2D) is the resistance of target tissue to insulin. Pathogenesis of T2D involves abnormalities in both insulin secretion and action (Ginsberg et al., 2000; Saltiel et al., 2001). Various in vitro and in vivo based experimental studies suggest that NF-κB activation is a key event early in the pathobiology of diabetes (Sandip et al., 2009). NF-κB induces iNOX mediated insulin resistance through insulin receptor substrate (IRS), followed by down-regulation of glucose transporter type-4 (GLUT-4). Alzheimer’s, a chronic and progressive neurodegenerative disease, is a leading cause of dementia in older people (Revadiger et al., 2014), which is characterized by the extracellular deposition of β-Amyloid (Aβ) aggregates (senile plaques) and intra-neuronal neurofibrillary tangles (hyper-phosphorylated tau proteins) and loss of neurons in the brain (Tamagno et al., 2008). Numerous studies have shown that Aβ or secreted form of the amyloid precursor protein (APP) induces up-regulation of NF-κB. Aβ and tau can undergo a non-enzymatic glycation and forms advanced glycation end-products (AGEs). These AGEs bind to a receptor of AGEs (RAGEs) and can trigger NF-κB dependent gene transcription (Vitek et al., 1994).
Tumorigenesis is a multistep process that can be activated by any of the various environmental carcinogens, inflammatory agents, and tumor promoters, which are known to modulate the transcriptional factors, pro-apoptotic proteins, protein kinases, cell cycle proteins, Cox-2, and growth factors (Aggarwal et al., 2004). A number of constitutively activated signaling pathways play a critical role in the survival and growth of cancer. These include NF-κB, phosphoinositide 3-kinase (PI3K)/protein kinase B and Janus kinase/signal transducer and activator of transcription survival pathway. NF-κB is widely recognized as the key positive regulator of cancer cell proliferation and cell survival via many pro-survival and anti-apoptotic genes (Feinman et al., 1999).
Constitutively activated NF-κB in chronic inflammatory patients has been found to be the critical linkage with a wide variety of human diseases, including asthma, atherosclerosis, AIDS, Alzheimer’s, Parkinson’s disease, diabetes (Sandip et al., 2009), myeloma (Feinman et al., 1999), acute myelogenous leukemia (Griffin et al., 2001), acute lymphocyte leukemia (Kordes et al., 2000), chronic myelogenous leukemia (Baron et al., 2002), and prostate (Palayoor et al., 1999), breast cancers (Nakshatri et al., 1997), rheumatoid arthritis, and osteoporosis which belong to autoimmune/inflammatory diseases (Gupta et al., 2010).
Many studies have reported that the compounds derived from plant source exert their anti-inflammatory and anticancer, anti-diabetic, and neuroprotective role through the suppression of NF-κB. NF-κB acts as a crossroad of many signaling pathways. Molecular regulation of NF-κB will give a great platform to exploit the transcriptional factor as a therapeutic target.
Present study is designed to evaluate the pharmacological properties of Benzo (f) chromen-3-one, a plant compound present in A. vulgaris (Sandeep et al., 2018) as a modulator in NF-κB pathway, a key regulatory molecule chronically activated in various ailments.
METHODOLOGY
Benzo (f) chromen-3-one, a plant compound present in A. vulgaris was selected for pharmacophore analysis. Benzo (f) chromen-3-one analogs were designed by modifying the pharmacophore moiety. Structures of Benzo (f) chromen-3-one were retrieved from PubChem database (CID-540269), and its analogs were drawn and analyzed using ChemDraw Ultra V6.0. 3D. Structures were energy minimized in Arguslab tool using universal force field with 1,000 steps and then converted to Autodock Pdbqt format in PyRx. Molecular properties of the compounds were analyzed using organic chemistry portal tool, Molinspiration (http://www.molinspiration.com/cgi-bin/properties). The Benzo (f) chromen-3-one molecule was studied using SWISS ADME tool. Three-dimensional X-ray crystal structure of NF-κB molecule (PDB ID: 3GUT) was retrieved from PDB (www.rcsb.org/pdb). Water molecules and heteroatoms present in the crystal structures were removed and partial charges were added to hetero groups and hydrogen atoms were added and energy minimized in Swiss PDB viewer (1223).
Chemical structures of Benzo (f) chromen-3-one and its analogs were subjected for virtual screening against NF-κB protein using A Lamarckian genetic algorithm in Autodock. Protein–ligand interactive visualization and analysis was carried out in Pymol viewer 1.5.4.
Protein network linked to inflammation, diabetes, Alzheimer’s, and cancer were analyzed to unfold the NF-κB mediated interacting mechanism using STRING version 10.0 (search tool for retrieval of interacting gene available at http://string-db.org (Szklarczyk et al., 2011). We used this tool to query, retrieve, and analyze the protein interaction network of PI3K, breast cancer anti-estrogen resistance protein 3 (BCAR3), beta-site APP-cleaving enzyme-1 (BACE1), NF-κB repressing factor (NKRF), and prostaglandin-endoperoxide synthase 2 (PTGS2) confined to those of Homo sapiens. From the home page, multiple protein search option is selected and query protein names were used to obtain interaction networks. An initial network was built with the selected proteins. Data settings were modified as zero proteins as first shell interactions and 10 proteins for second shell interactions to minimize the complication in the interaction network. We performed NKRF and NF-κB interaction as a second cluster with three proteins in the second shell. Finally, two clusters were linked to form a clear view. Prediction method was selected for our analysis which includes gene fusion, database, text mining, co-expression, co-occurrence, and neighborhood. Evidence view display option was selected for color variation representing the source of interaction data. KEGG pathways were retrieved for inflammation, diabetes, Alzheimer’s and cancer.
RESULT AND DISCUSSION
Based on the paucity of scientific studies on Benzo (f) chromen-3-one , the present work is carried out to evaluate the role of this compound on the NF-κB pathway during inflammatory mediated pathogenesis through in silico approach. Benzo (f) chromen-3-one and its analogs are shown in the Table 1. Benzo (f) chromen-3-one and its analogs were docked against NF-κB which showed greater binding affinity scores. Binding affinity scores of Benzo (f) chromen-3-one and its analogs with molecular properties were listed in the Table 2. Compound binds to the DNA binding domain of NF-κB molecules with a binding affinity of −7.8 kcal/mol. Biochemical studies on conserved DNA binding interactions studies by James et al. (2009) clearly demonstrated the protein DNA interactions of NF-κB (Fig. 3). Benzo (f) chromen-3-one shows bonding interactions with Arg 605 (Arg NH2–1HH2—O22 of benzochromen-3-one with bond length of 2.8, Ao, Arg NH2–2HH2—O20 of benzochromen-3-one with bond length of 2.5 Ao) and Asp217 (Asp OG1–HG1—O22 of benzochromen-3-one with bond length of 2.3Ao) and also shows hydrophobic interaction with the surrounding amino acids Arg33, Asn186, Arg 187, Pro 189, Glu 193, Ala 192, and Arg 605. Among the surrounding molecules Ala 33 and Arg187 of Rel-A, Gln606 of P50 are involved in hydrophobic interaction with DNA. Interaction image is shown in the Figure 2.
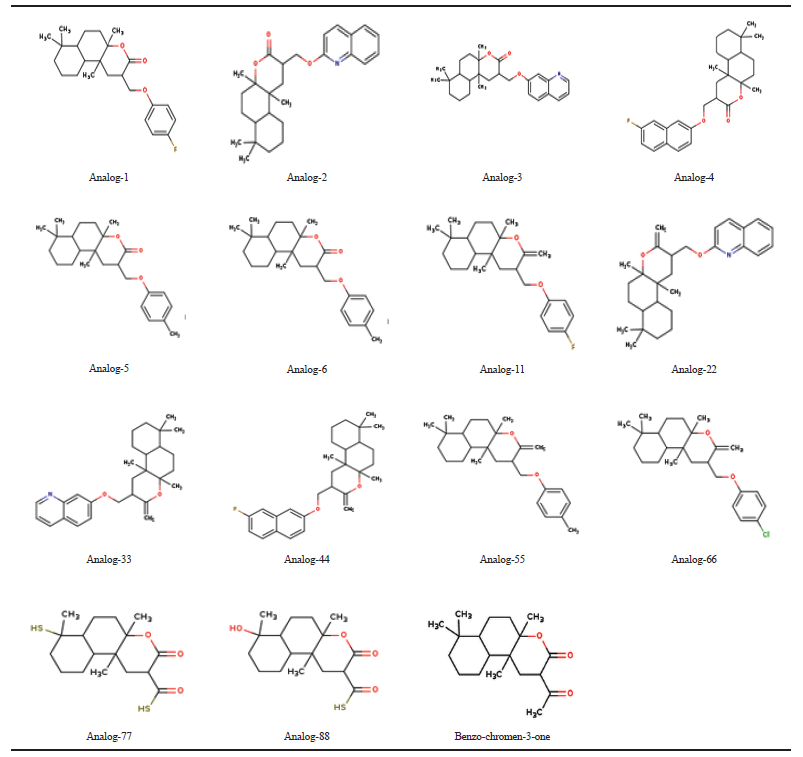 | Table 1. Benzo (f) chromeno-3-one analogs. [Click here to view] |
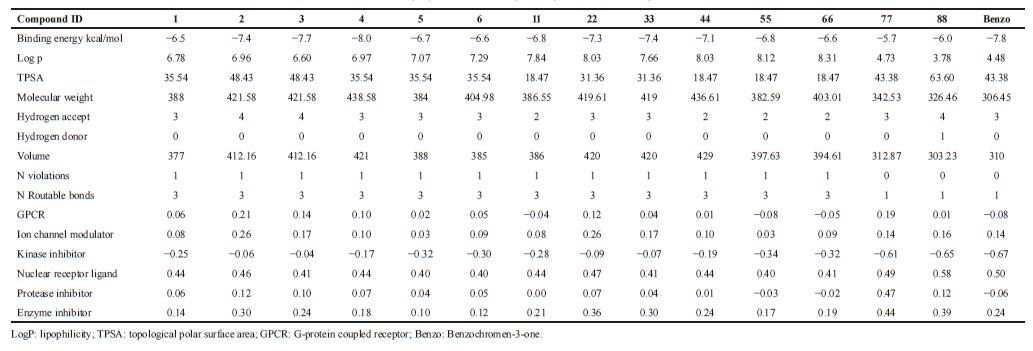 | Table 2. Molecular properties of the compounds predicted in Molinspiration server. [Click here to view] |
Molecular properties of the compound were analyzed using Molinspiration and Swiss ADME servers. The result shows that the compound satisfies the Lipinski rule of five with no violations having a molecular weight of 306.44 Daltons showing one rotatable hydrogen bond and three hydrogen bond acceptors (Table 3). Bio-activity predictions indicate that the compound acts as a nuclear receptor inhibitor, and these results correlate with molecular docking studies which showed the greater binding affinity of the compound with NF-κB. SWISS ADME results indicate that the gastrointestinal absorption of the compound is high, which indicates that the compound can be absorbed easily when taken in oral route. All the analogs showed greater log P value, which indicates molecular hydrophobicity. Hydrophobicity affects drug absorption, bioavailability, hydrophobic drug-receptor interactions, metabolism of molecules, as well as their toxicity. Log P has become also a key parameter in studies of the environmental fate of chemicals. Whereas analog-77 showed poor solubility and analog-88 showed three violations in SWISS ADME result.
Protein interaction network developed in STRING Database, based on gene fusion, database, text mining, co-expression, co-occurrence, and neighborhood, showed the connection between signaling mechanisms involved in inflammation, Alzheimers, cancer, and diabetes (Fig. 1). PTGS2 (prostaglandin G/H synthase and cyclooxygenase) mediates the formation of prostaglandins from arachidonate and is involved in inflammatory mechanism in the arachidonic acid signaling cascade. IGF1R and IRS are implicated in insulin signaling essential for translocation of GLUT-4. Insulin resistance mediated by NF-κB through JNK/iNOS inhibits the activation of PI3K, phosphorylation of AKT by PI3K, thereby inhibiting the translocation of glucose transporter-4. BACE and APP are known to play a major role in neurological inflammation mediated through NF-κB activated iNOS. BCAR3 gene is implicated in breast cancer, which acts as an adapter protein and couple activated growth factor receptors to a signaling pathway that regulates the proliferation of breast cancer cells. When overexpressed, it confers anti-estrogen resistance in breast cancer cell lines. Recent studies on Tumor promotor 53 (TP53), a tumor suppressor gene indicates that mutant P53 elevates the expression of P52 NF-κB by acetylation of Histones. Mutant P53 is much more involved in many cancers (Bastian et al., 2013). C-JUN protects the cell from UV radiation-induced apoptosis. C-Jun co-operates with NF-κB to prevent TNF-alpha induced apoptosis (Ron et al., 1999).
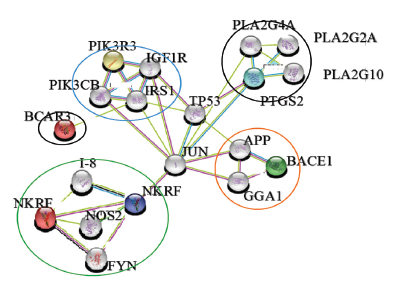 | Figure 1. Molecular interactive network showing connecting link between various diseases. (a) blue—diabetes, (b) pink—inflammation, (c) orange—Alzheimer’s, (d) black—cancer, and (e) green-NF-κB transcriptional factors. [Click here to view] |
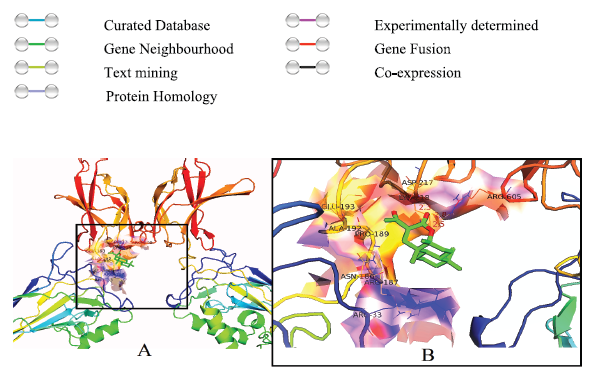 | Figure 2. Molecular docking studies of Benzo (f) chromen-3-one with NF-κB. (A) Showing the binding site of benzo (f) chromen-3-one to NF-κB; (B) visualization of interacting amino acids with the compound. [Click here to view] |
When an injury occurs, sensory information rapidly alerts the brain and begins the complex sequence of events to restrain homeostasis. Cytokines are released within a fraction of seconds after an injury. Cytokines such as gamma interferons, IL-1 and IL-6, and TNF are released immediately after an injury, enter into the bloodstream, and send a signal to the brain. As a counteraction from the brain, wound healing and inflammation mechanism are activated. In the early stage, many chemical mediators of inflammation are produced resulting in arachidonic acid cascade and the release of growth factors, such as platelet-derived growth factor, platelet factor 4, insulin-like growth factor, and transforming growth factor.
At molecular level, inflammation is regulated by numerous molecules and factors, adhesion molecules, vascular cell adhesion molecules, endothelial leucocyte adhesion molecule, chemokines, cytokines, signal transducer and activator of transcription, proinflammatory enzymes, vascular endothelial growth factor, and proinflammatory transcriptional factor such as NF-κB (Aggarwal et al., 2004). Among these mediators, NF-κB is the central regulator of inflammation. NF-κB regulates the expression of almost 500 different genes, including enzymes, cytokines, adhesion molecules, cell cycle regulatory molecules, and angiogenic factors, which are implicated in inflammation-related responses (Shih et al., 2015).
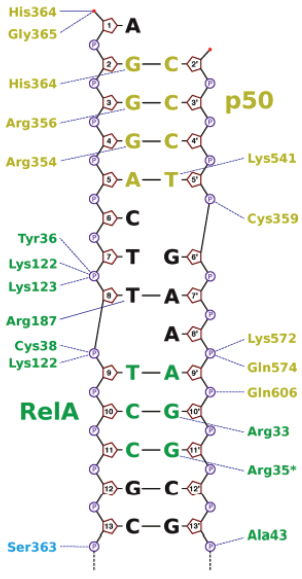 | Figure 3. Interaction image of DNA with amino acids in NF-κB. [Click here to view] |
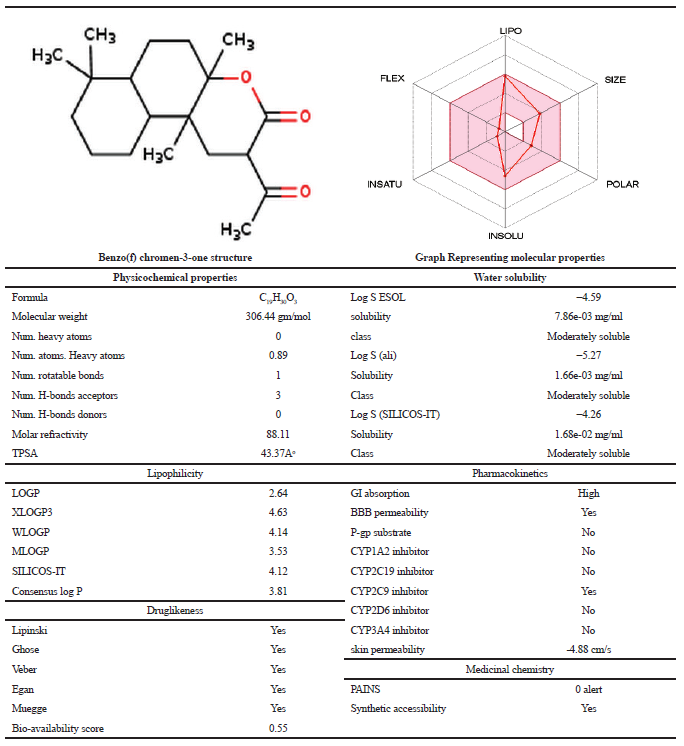 | Table 3. Molecular properties of Benzo (f) chromen 3 one. [Click here to view] |
Hyperinsulinemia and obesity are the predominant complications in T2D (Herling et al., 2011). T2D and obesity are proinflammatory conditions that are activated with increased production of inflammatory cytokines, such as IL-6 and TNF-α by adipose tissue (Eltzschig et al., 2011). TNF is an NF-κB regulated product as well as a potent activator of NF-κB. NF-κB has become a chief suspect in the development of insulin resistance and T2D (Kowluru et al., 2003). Many researchers have highlighted the role of NF-κB in the pathogenesis of insulin resistance and T2D (Arkan et al., 2005). NF-κB induces iNOS mediated insulin resistance predominantly through the serine phosphorylation of insulin receptor substrate-1 (IRS1), thereby contributing to insulin resistance and down-regulation of GLUT-4 expression (Kahn et al., 2000), which is clearly shown in the KEGG pathway analysis. Conditional and specific NF-κB inhibition protects pancreatic Beta cells from cytokine-induced apoptosis in vitro and in vivo (Eldor et al., 2006). It was hypothesized that the NF-κB attenuation would show a protective effect in diabetic patients with insulin resistance.
Alzheimer’s disease (AD) has been explained by three hypothesis, such as amyloid β hypothesis, tangle hypothesis, and cholinergic hypothesis (Tougu et al., 2001). Amyloid beta peptide plays a central role in the induction of inflammation in AD. Accumulation of amyloid beta peptides results in increased levels of inflammatory molecules, such as cytokines, chemokines, and competent cells produced by glial cells further leading to neuronal damage (Griffin et al., 1998). TNF-α is one of the most predominant pro-inflammatory cytokines which is significantly increased in ’AD and plays a crucial role in increased levels of cytokine cascade during inflammation (Fillit et al., 1991). As NF-κB is primarily activated in inflammation, it also plays a critical role in the regulation of neuroinflammation associated with disease pathogenesis. The activation of NF-κB transduction factor is responsible for neurogenesis, neuritogenesis, synaptic plasticity, learning, and memory. NF-κB was known to exist in the cytosol in an inactive form in all cell types including the nervous system.
Recent evidences have shown that activation of NF-κB drives systemic and brain aging process in mice. KEGG pathway analysis clearly visualizes the TNF-alpha mediated NF-κB activation. NF-κB induces iNOX mediated neurofilament damage further gives rise to the impairment of axonal transformation and finally leads to cell death. TNF-α performs its biological function via two distinct receptors TNF-R1 and TNF-R2. Simultaneous activation of both TNF-α receptors leads to the activation of NF-κB, which have a binding site for both AβPP and BACE gene 9 (Grilli et al., 1996). Elevated expression of both AβPP and BACE expression will ultimately lead to increased Aβ production, which in turn, activates glial cells and enhance neuronal inflammation process. Research evidence on the brain of Alzheimer’s patients showed that the activated NF-κB was found predominantly in neurons and glial cells in Aβ plaque regions (Kaltschmidt et al., 1997). Since the compound, benzo (f) Chromen-3-one showed greater binding affinity with NF-κB, it crosses blood brain barrier and inhibits constitutive activation of NF-κB in the brain. These results hypothesize the protective effect of a compound against NF-κB mediated neuronal inflammation in Alzheimer’s and Parkinson’s diseases.
NF-κB activity has been constitutively elevated in many human tumors, melanomas (Amiri et al., 2005), breast (Sovak et al., 1997), prostate, ovarian, pancreatic (Sclabas et al., 2005), colon (Kojima et al., 2004), thyroid (Pacifico et al., 2004), carcinomas, and glioblastoma. NF-κB functions as an inhibitor of apoptosis and hence inhibition of NF-κB by genetic or chemical inhibitors induces apoptosis in various tumors. TNF-α mediates the development and advancement of many tumors by activating nuclear factor NF-κB (Pikarsky et al., 2004), and hence NF-κB inhibitors can be used for the treatment of many cancers. It is reasonable to conclude that Benzo (f) chromen-3-one could possibly inhibit the transcription activity of NF-κB as it is well placed in the DNA binding domain, which may affect the binding of NF-κB to the DNA. Isolation, characterization, in vivo and in vitro studies of benzo (f) chromen-3-one is in progress.
CONCLUSION
Our computational analysis provides a clear evidence for the inhibitory ability of plant compound present in A. vulgaris to alter the NF-κB signaling pathway. Docking studies substantiate the hypothesis that Benzo (f) chromen-3-one has a potentiality to inhibit the NF-κB, by binding to the DNA binding domain of NF-κB that would possibly disrupt the stability of NF-κB—DNA complex formation in the transcription of genes. Based on in silico approach, benzo (f) chromen-3-one is suggested for in vitro and in vivo studies as a potential drug candidate for inflammation-induced disease pathogenesis.
REFERENCES
Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell, 2004; 6(3):203–8.
Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev, 2005; 24(2):301–13.
Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med, 2005; 11(2):191–8.
Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol, 1996; 14:649–83.
Baron F, Turhan AG, Giron-Michel J, Azzarone B, Bentires-Alj M, Bours V, Bourhis JH, Chouaib S, Caignard A. Leukemic target susceptibility to natural killer cytotoxicity: relationship with BCR-ABL expression. Blood, 2002; 99(6):2107–13.
Bastian H, Johannes AS. The complexity of NF-κB signalling in inflammation and cancer. Mol Cancer, 2013; 12:86.
Collins T, Cybulsky MI. NF-kappa B: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest, .2001; 107(3):255–64.
Eldor R, Yeffet A, Baum K, Doviner V, Amar D, Ben-Neriah Y, Christofori G, Peled A, Carel JC, Boitard C, Klein T, Serup P, Eizirik DL, Melloul D. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc Natl Acad Sci USA, 2006; 103(13):5072–7.
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med, 2011; 364(7):656–65.
Feinman R, Koury J, Thames M, Barlogie B, Epstein J, Siegel DS. Role of NF-κB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood, 1999; 93(9):3044–52.
Ferrero S, Colombo BM. Differences between plasma and serum vascular endothelial growth factor. Am J Cardiol, 2006; 98(3):424–5.
Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett, 1991; 129(2):318–20.
Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol, 1998; 16:225–60.
Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest, 2000; 106(4):453–8.
Griffin JD. Leukemia stem cells and constitutive activation of NFκB. Blood, 2001; 98:2291.
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol, 1998; 8(1):65–72.
Grilli M, Goffi F, Memo M, Spano P. Interleukin-1 beta and glutamate activate the NF-kappa B/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J Biol Chem, 1996; 271:15002–7.
Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta, 2010; 1799(10–12):775–87.
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 1997; 18:2714–23.
Herling A, König M, Bulik S, Holzhütter HG. Enzymatic features of the glucose metabolism in tumor cells. FEBS J, 2011; 278(14):2436–59.
James CS, Amy O, Aidong H, Darren LB, Lin C. Structural basis of HIV-1 activation by NF-kappa B - a higher-order complex of p50:RelA bound to the HIV-1 LTR. J Mol Biol, 2009; 393(1):98–112.
Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest, 2000; 106(4):473–81.
Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappa B is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997; 94(6):2642–7.
Karin M, Greten FR. NF-kappa B: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol, 2005; 5(10):749–59.
Kojima M, Morisaki T, Sasaki N, Nakano K, Mibu R, Tanaka M, Katano M. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res, 2004; 24(2B):675–81.
Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, Scheidereit C. Transcription factor NF-κB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia, 2000; 14:399–402.
Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res, 2003; 37(11):1169–80.
Maslinska D, Gajewski M. Some aspects of the inflammatory process. Folia Neuropathol, 1998; 36:199–204.
Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr. Constitutive activation of NF-kappa B during progression of breast cancer to hormone-independent growth. Mol Cell Biol, 1997; 17(7):3629–39.
Pacifico F, Mauro C, Barone C, Crescenzi E, Mellone S, Monaco M, Chiappetta G, Terrazzano G, Liguoro D, Vito P, Consiglio E, Formisano S, Leonardi A. Oncogenic and anti-apoptotic activity of NF-kappa B in human thyroid carcinomas. J Biol Chem, 2004; 279(52):54610–9.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene, 1999; 18(49):6853–66.
Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Constitutive activation of IkappaB kinase alpha and NF-kappa B in prostate cancer cells is inhibited by ibuprofen. Oncogene, 1999; 18(51):7389–94.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappa B functions as a tumour promoter in inflammation-associated cancer. Nature, 2004; 431(7007):461–6.
Ron W, Randall SJ, Connie M. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J, 1999; 18(1):188–97.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 2001; 414(6865):799–806.
Sandeep Kumar G, Ramaiah CV, Rajendra W. Qualitative and quantitative phytochemical profile and In vitro anti-oxidant activity of methanolic extract of Artemisia vulgaris. Pharma Innov J, 2018; 7(9):348–54.
Sandip P, Dev S. Role of NF-κB in the pathogenesis of diabetes and its associated complications. Pharmacol Rep 2009; 61:595–603.
Sclabas GM, Uwagawa T, Schmidt C, Hess KR, Evans DB, Abbruzzese JL, Chiao PJ. Nuclear factor kappa B activation is a potential target for preventing pancreatic carcinoma by aspirin. Cancer, 2005; 103(12):2485–90.
Shih RH, Wang CY, Yang CM. NF-kappa B signalling pathways in neurological inflammation: a mini review. Front Mol Neurosci, 2015; 8:77.
Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE. Aberrant nuclear factor-kappa B/Rel expression and the pathogenesis of breast cancer. J Clin Invest, 1997; 100(12):2952–60.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins globally integrated and scored. Nucleic Acids Res, 2011; 39(Database issue):D561–8.
Tak PP, Firestein GS. NF-kappa B: a key role in inflammatory diseases. J Clin Invest, 2001; 107(1):7–11.
Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo D-G, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the γ- and β-secretase cleavages of the β-amyloid precursor protein. J Neurochem, 2008; 104:683–95.
Tougu V. Acetylcholinesterase: mechanism of catalysis and inhibition. Curr Med Chem – CNS Agents, 2001; 1:155–70.
Revadigar V, Ghalib RM, Murugaiyah V, Embaby MA, Jawad A, Mehdi SH, Hashim R, Sulaiman O. Enzyme inhibitors involved in the treatment of Alzheimer’s disease. Drug Des Discov Alzheimer’s Dis, 2014:142–198
Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA, 1994; 91(11):4766–70.