
INTRODUCTION
In developed countries such as India, the United States, and those in Europe, Staphylococcus aureus strains that incorporate virulent and resistant genes are being recognized as a significant therapeutic concern. For decades, this has been an important issue for global health, impacting millions of people worldwide. The distinctive capability of the bacterial enzyme DNA gyrase to form negative supercoils in closed-circular DNA is noteworthy. Staphylococcus aureus generates negative supercoiling through the action of the DNA gyrase enzyme. Inhibition of the GyrB gene would prevent DNA supercoiling, thereby hindering S. aureus from replicating and stopping the infection from spreading. Since suppressing this enzyme is necessary for various biological processes, it serves as an effective antibacterial strategy [1]. The protein known as the DNA supercoiling factor, which interacts with eukaryotic topoisomerase II to create negative supercoils in DNA, was first identified in silkworms. These eukaryotic systems exhibit “passive” supercoiling, in contrast to the active supercoiling mediated by DNA gyrase. Passive supercoiling arises from mechanical influences, such as DNA replication, rather than enzymatic processes. In contrast, active supercoiling regulates DNA supercoiling through enzymes, particularly topoisomerases such as DNA gyrase. These enzymes can modulate the level of supercoiling, thereby affecting various DNA-related functions. Most antibacterial substances that specifically target DNA gyrase can be divided into two groups: quinolones and coumarins [2].
The discovery was made in 1880 by Scottish surgeon Alexander Ogston, who noticed bacterial colonies in the pus after a surgical incision. Initially, penicillin had a high success rate for treating S. aureus, but, by the end of the 1940s, penicillin resistance had sharply developed [3]. On blood agar plates, S. aureus is a stationary, spherical, golden-yellow Gram-positive bacterium. It reproduces asexually through binary fission, in which autolysin facilitates the daughter cells’ separation from the mother cell. Pre-existing, extremely effective resistance mechanisms to several antibiotics developed in antibiotic manufacturers or their rivals and were too readily acquired by pathogenic staphylococci through horizontal gene transfer, comprising genetic components that are migratory. These factors might have developed in antibiotic producers as a defense against substances that could hinder them or in their rivals [4].
In 1976, Menzel et al. [5] discovered the revolutionary breakthrough of DNA gyrase using Escherichia coli. DNA gyrase is an ATP-dependent enzyme that may add supercoils that are negative to closed-circular duplex DNA. Two classes of DNA synthesis inhibitors, quinolones and coumarins, had also been explored before this discovery [5]. Subsequent research has shown that DNA gyrase is the main target of quinolones (nalidixic acid, oxalinic acid, and ciprofloxacin) and coumarins (novobiocin, coumermycin, and clorobiocin). It was also shown that the genes mlA and mu, which previously had been linked with resistance to coumermycin or nalidixic acid, really encode two distinct proteins that function together to form gyrase. After that, they were dubbed gyrA and gyrB. They are located at 48 and 83 minutes on the E. coli K-12 chromosome, and mutations in these loci result in drug- and temperature-sensitive forms of gyrase. Furthermore, the gyrase enzyme has been discovered in several other species [6].
Because of the development of antibiotic resistance, the manufacture of toxins and enzymes, the ability to form biofilms, and the ability to evade immune responses, the existing treatments for Staphylococcal infections are not always effective. The rapid ascent of S. aureus to prominence as a dangerous human illness with global implications can be attributed to all of these factors. However, the inability to develop effective treatments for Gram-positive pathogen infections is due to biofilm development, which is made worse by the fact that S. aureus and highly resistant methicillin-resistant S. aureus both have different biofilm processes.
Rifampin is no longer advised for use as a monotherapy for immediate bacterial skin and skin structure infections due to the rapid acquisition of resistance. The efficacy of tetracyclines and TMP-SMX against CAMRSA is limited, and the presence of beta-lactam drugs increases the development of clindamycin resistance. Tetracyclines could become photosensitized as well. However, minocycline and doxycycline also have a limited effect on adults and adolescents due to their staining of dental enamel. All medicinal substances have historically originated in the natural world, with higher plants accounting for many of these sources. As of right now, higher plants are important historical sources of new compounds that have direct medical applications, can be used as model compounds to optimize and modify synthetic or semisynthetic structures or can be used as pharmacological or biochemical probes. Among the plant-derived compounds that have recently been developed are the anticancer medications Taxol and camptothecin, the Chinese antimalarial therapy artemisinin, and the East Indian Ayurvedic therapeutic forskolin. These and many other examples show the value of secondary metabolites produced from plants as prospective molecules for the development of modern medicine. Furthermore, since conventional treatments are chemical-based, this study concentrates on a naturally occurring plant-based medication for staphylococcal infections. This medication targets the DNA gyrB gene, which causes the DNA to supercoil, halting the transmission of infection and curing disorders. Using systems biology approaches and molecular docking simulations, bioactive compounds from Vitex negundo, Euphorbia hirta, and Solanum nigrum will be tested for their potential to interact with key proteins implicated in bacterial pathogenicity, specifically the DNA gyrB gene. Molecular dynamics simulations can revolutionize therapeutic strategies by providing a more comprehensive evaluation of the pharmacological properties of lead compounds [7].
MATERIALS AND METHODS
Protein–protein interaction (PPI) string analysis
Using the biology repository and web tool, STRING server has been used for the pre-computed gene or protein, and the interaction between the various interconnected proteins is predicted. The tool asses the functional enrichment for the given lists of proteins or genes using functional classification systems such as GO, Pfam, and KEGG. Protein–protein interactions are annotated with “scores,” or confidence marks. The scale runs from 0 to 1, where 1 signifies the highest degree of certainty [8].
Gene interaction network construction
Network-based methods overlay genes or proteins that appear insignificant in a large-scale network environment to provide an analytical framework and a comprehensive presentation of genomic data from high-throughput studies. By using network modules, the number of dimensions in the data is reduced and statistical techniques are more successful. A variety of supplemental data sources are used to construct the functional interface network, and human-curated network databases are used to extract the network’s functional links. Cytoscape is the most useful tool for studying and visualizing biological networks. This has made it possible to quickly and cheaply adopt cutting-edge network biology techniques, such as dialect libraries, carefully customized studies, Cytoscape process integrations, and best-in-class tools [9].
Virtual screening of phytochemicals
Determining the concentration of different bioactive components in plants is made easier with the use of the GC-MS method, which was employed for the analysis of the produced plant extracts. Medicinal plants, which form the foundation of traditional medicine, have been the subject of much contemporary pharmacological research. This has been made feasible by the realization of the medicinal plants’ significance as sources of novel therapeutic compounds and lead molecules for pharmaceutical development [10].
The GC spectrum study indicates that the extracts of E. hirta, V. negundo, and S. nigrum had the highest GC values for various compounds in their GC fraction. The lead compounds retrieved from the GCMS analysis are displayed in Table 1. The M/Z ratio, molecular formula, and retention time in minutes of the retrieved compounds are given in Table 2. Additionally, each detected compound’s mass spectrum and chromatogram graph are included in the supplemental file separately.
 | Table 1. Libraries retrieved from the GCMS analysis of extracts of E. hirta, V. negundo, and S. nigrum. [Click here to view] |
 | Table 2. Indicating the name of the compounds, M/Z ratio, molecular formula, and retention time, of E. hirta, V. negundo, and S. nigrum extracts as shown by the GC- MS analysis. [Click here to view] |
Molecular docking
Molecular docking is being extensively explored as a lead-discovery method as the structures of an increasing number of proteins and nucleic acids are known. As more experimentally described protein structures are found, the number of proteins that can be docked against homology-modeled targets rises. As more docking experiments are carried out, the degree of accuracy and “drug-likeness” of docking hits are being examined. With upgrades to relevant search algorithms and enhancements to the scoring function, the molecular docking approach will become a reliable drug-design tool that incorporates significant volumes of biological data [11]. Autodock Vina (MGLtools 1.5.7) was used for active site identification, ligand preparation, molecular docking studies, and energy minimization performed by NAMD software. Protein DNA GyrB, derived from the PDB website with PDB ID 5D7D, was docked against the lead compounds for further studies.
Preparation of protein as PDBQT files
The docking workspace is now located in the preference folder. The prepared protein has been brought into the Auto Dock 1.5.6 workspace. Estimating the protein’s Gasteiger charges after simply adding polar hydrogen atoms is the initial step. After that, Kollman charges were introduced. The protein was stored in the PDBQT file format. Next, the ligand was added, and the torsion tree’s root was selected. Additionally, the ligand was stored in the PDBQT format. To carry out the computational procedure, the ligand and protein were loaded into the computing environment in the PDBQT format.
Grid parameters
To ascertain whether the ligand was successfully bound to the protein’s active site, the active site residues identified using pymol visualization software with the help of protein sequence from PDB (PDB ID: 5D7D) mentioned in Table 3 were applied. Assigning the grid parameters, which direct the ligand to the protein’s required location, is the most important stage in the molecular docking process. The area of the protein where docking will take place is indicated by the grid box position. During docking, no area outside the box will be investigated. The docking’s perimeter was established using the grid box. The grid box should encompass the entire macromolecule if you want to examine every surface possible without knowing the protein’s active site. The grid box’s center and measurements were selected so as to enable the protein–ligand complex to fit inside of it. Grid spacing was set to 0.647 Å by default. The values of x = 22.757, y = 4.436, and z = 33.904 were fixed for the center grid box, with offset values of −8.743, −36.064, and 6.154, respectively. The dimensions of x, y, and z were assigned 84 × 108 × 74 grid points. The corresponding maps had a total of 67,1533 grid points. Compounds are docked to binding sites with known structures using genetic algorithms. During the docking procedure, the genetic algorithm technique is used to decrease the energy of intermolecular interactions. The docking run of the genetic algorithm was 10. These characteristics surrounded the protein’s whole three-dimensional active site. The output was stored in the grid parameter file (GPF) format by the grid parameter.
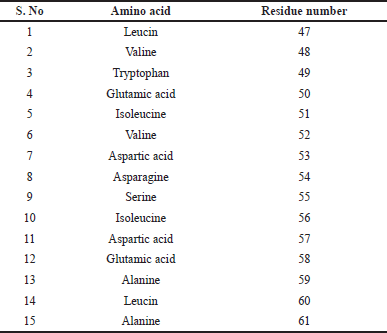 | Table 3. Determination of active site of the protein. [Click here to view] |
Autogrid and autodock run
The grid log file was prepared by the Auto Grid application using the GPF file as input after the protein’s active site was predicted. Both the grid-run and the genetic approach were used. Docking parameter file format (DPF) was used to construct the output file. Following the docking log file (DLG) preparation, the Autodock runtime and DPF data were utilized as input for additional docking analysis. The final DLG file contained the inhibitory constants and the 10 most significant free-binding energies for each experiment. The complex with the lowest binding energy was saved for further study in the PDBQT file format after data processing, binding energy sorting, and PDB format description.
Protein–ligand interaction profiler (PLIP)
Software called the PLIP aids in confirming both the kind and degree of interaction between a protein and ligand. The amino acid and ligand bond lengths as well as the residues are extracted from the PLIP. The precise interaction types and bond lengths of the docked complex are provided when the PDB file of the protein–ligand docked complex is used as the input file.
In Silico ADME analysis and pharmacokinetic study
This study presents the free Swiss ADME server, a web-based tool to assess the ADME properties of phytoconstituents. Understanding the biological and pharmacological characteristics of the plant can be aided by this information. Swiss ADME uses an analytical method based on the Lipinski rule of 5 to assess the bioavailability, drug-like characteristics, and biopharmaceutical compatibility of the bioactive compounds that are collected. The study of a drug’s distribution, metabolism, removal, and absorption is known as pharmacokinetics. Drug distribution, biotransformation, excretion, and absorption are the main times when drug–drug interactions happen. The number of binding proteins has a basic influence on distribution, and many medications alter protein binding. The medication is transformed into active or inactive metabolites as it moves through the liver. This process is called biotransformation, and it has a big impact on how well medications work [12].
Molecular dynamics
Molecular biology and drug discovery domains have seen a notable surge in the application of molecular dynamics (MD) simulations in the past few years. These simulations faithfully capture the atomic-level behavior of proteins and other biomolecules at incredibly fine temporal resolutions. Simulation computations have been useful in the investigation of protein–biomolecule interactions, in the synthesis of novel proteins, peptides, and small molecules, and in determining the structural causes of disease [13]. A comparable molecular dynamics program called NAMD version 2.14 is intended for efficient modeling of large biomolecular systems. The file formats, future CHARMM functionalities, and limits all function well with NAMD. When combined with NAMD, the molecular graphics program visual molecular dynamics (VMD) version 1.9.4 offers a thorough modeling environment. Because of its other features, VMD can provide functionality that NAMD cannot. VMD provides a unique environment for examining data from NAMD simulations [14].
NAMD simulations of MD require a number of consecutive stages. First, the biomolecular system is configured, and formatting and parameterization are appropriately applied. The force field that most closely resembles the properties of the system is chosen. It is necessary to build input files in order to supply simulation parameters such as pressure, temperature, and boundary conditions. These files include coordinate, parameter, topology, and configuration files. The next stage in addressing steric conflicts and maintaining the structure is energy minimization. After that, the system is heated and maintained at the target temperature for the duration of equilibration, after which any positional restrictions are progressively lifted. Production begins for the specified amount of time, and MD simulation begins and integrates dynamics using chosen interaction algorithms and periodic boundary conditions. Understanding the generated trajectory data’s dynamic behavior and structural features comes via analysis. Data are refined for analysis and visualization through postprocessing, which results in methodology, results reporting, and documentation. These steps enable a full investigation of biomolecular systems using NAMD, leading to significant new insights into their interactions and behavior [15].
MD simulations are used in the MMPBSA method via Ambeer Tools (version 23) to find the binding free energy in molecular systems. Next, snapshots are taken in order to estimate the solvation-free energy and conformational entropy. By offering perspectives on how molecules interact using a comparison of the total free energy, binding energy, van der Waals energy, electrostatic energy, and polar solvation of complex, receptor, and ligand states, it aids in ligand selection and therapeutic focus [14].
System preparation with CHARMM
Due to various features for analyzing and altering atomic orientations and dynamics courses, the CHARMM tool is a popular package for analyzing macromolecular mechanics and dynamics. The creation of runs of a molecule’s dynamical trajectory and the minimization of a given structure are the two most basic techniques in simulation. The system must first be built up, which is done using CHARMM. This entails choosing a force field, assembling a simulation box, and carrying out NVT-NPT equilibration, charge neutralization, solvation, and energy reduction. We will use NAMD to model fabrication and equilibration in the second section. One popular atomic-level force field for simulating biological systems, especially proteins and nucleic acids, is CHARMM22. The force field is engineered to precisely simulate the behaviors of biological molecules, encompassing electrostatic, van der Waals, and hydrogen bonding interactions. In the NAMD molecular simulation procedure, the CHARMM22 force field was employed. Local interaction terms in this force field include pairwise interactions such as van der Waals and electrostatic forces, as well as bound interactions between two, three, and four atoms.
Equilibration and production run
Once the system has been properly configured with the CHARMM GUI, the output is generated in the (.tar) format. The files production.inp and equilibrium.inp are in this format. The (.inp) files must be run on the NAMD platform for the required number of picoseconds. You must download and run the equilibrium and production input data on your workstations because of the substantial processing power needed for extended simulations. The result can be seen and understood using the VMD tool. Using conventional simulation parameters, NAMD molecular simulation operates as follows. The quantity of simulation timesteps must be completed. It is allowed to use an integer larger than 0 (Numsteps × timestep) that is the entire simulation time. The timestep size should be used to integrate each simulation step. The unit of measurement is femtoseconds. In CHARMM-GUI, KCl is employed as a neutralizing species to neutralize a system. The physiological ion concentration of 0.15 M is the default ion concentration. Protein–ligand complex architectures’ docking findings are enhanced by the use of KCl ions.
RESULTS AND DISCUSSION
String analysis of protein–protein interaction
Predicting interactions between proteins for the pre-calculated gene or protein is done using the biology database and the web application STRING server. It uses functional classification systems such as GO, Pfam, and KEGG to evaluate the functional enrichment of the given lists of proteins or genes. Figure 1’s protein–protein interaction is marked with “scores” or confidence markers. All ratings are on a scale from 0 to 1, where 1 is the highest level of confidence. The program provides a group of genes known as nodes based on the greatest confidence level (0.9) after string interactive network analysis. The nodes in this string network indicate that the gyrB protein has a greater interaction rate than all other proteins in Table 4, which illustrates how the protein interacts with other proteins in the database. Furthermore, the molecular interaction network on these nodes is constructed using Cytoscape.
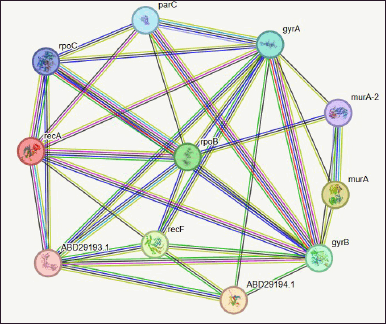 | Figure 1. Network of protein–protein interaction from string database for the proteins retrieved from NCBI. [Click here to view] |
 | Table 4. Nodes provided by the string interactive network analysis. [Click here to view] |
Pathway analysis—gene ontology enrichment
The computer representation of current discoveries on the functions of both coding and noncoding genes is called gene ontology. Gene ontologies, or functional enrichment of genes reconstructed as nodes from string databases, are shown in Table 5. Each ontology explains a particular feature of the functionality of a gene or gene product, in addition to the relationships between the ideas. A gene that is linked to a term implies that it is linked to all of the term’s parents as well. Further focusing the search on genes that interact within an organism or are comparable between two organisms may also be helpful.
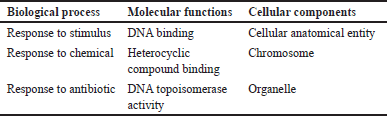 | Table 5. The gene ontologies (functional enrichment) of genes retrieved from the string database. [Click here to view] |
Gene network construction
Gene network interaction was calculated to identify the functional relationship among the genes, which is calculated in the form of rank. In this study, we have calculated the MMC, degree method, closeness, and betweenness method. The GyrB gene has the highest rank of 1 in all the four methods, as shown in Tables 6–9.
 | Table 6. Ranking of genes by maximal clique centrality (MCC) method. [Click here to view] |
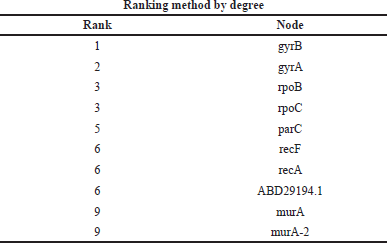 | Table 7. Ranking of genes by a degree method. [Click here to view] |
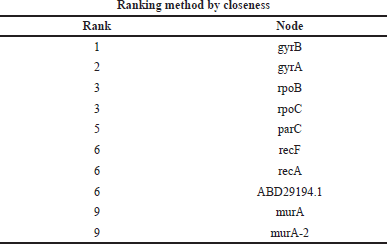 | Table 8. Ranking of genes by the closeness method. [Click here to view] |
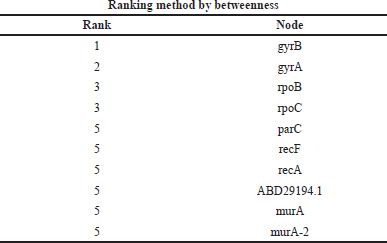 | Table 9. Ranking of genes by the betweenness method. [Click here to view] |
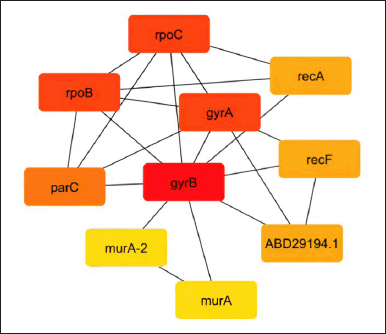
As shown in Figures 2–5, the topological algorithms are used in this study to combine the global techniques such as maximum clique centrality (MCC), degree, and closeness with local techniques like betweenness.
 | Figure 2. Network construction by the maximal clique centrality (MCC) method. [Click here to view] |
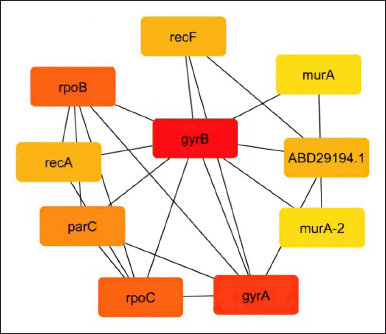 | Figure 3. Network construction by the degree method. [Click here to view] |
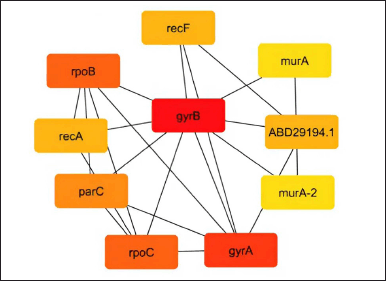 | Figure 4. Network construction by the closeness method. [Click here to view] |
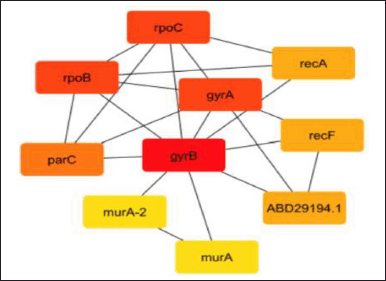 | Figure 5. Network construction by the betweenness method. [Click here to view] |
Molecular docking
The affinity between a protein and its ligand increases with negative binding energy. Docking forecasts the optimal configuration of one molecule for another when a target (ligand) and a protein create a stable complex together. Once the optimal orientation has been established, factors, such as binding energy, link stability, or binding affinity, among the two molecules can be used to forecast which ligand will be chosen for simulation experiments. The binding energies of ligands against the DNA gyrB gene are shown in Table 10.
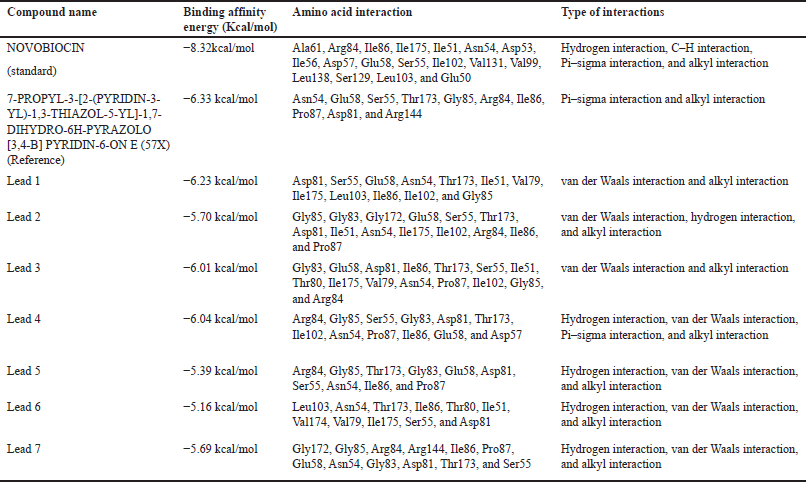 | Table 10. Binding energies and residue interactions between DNA gyrB gene and ligands. [Click here to view] |
Commercial drugs such as novobiocin are used for managing bacterial infections, especially those caused by Gram-positive bacteria. It is primarily used to treat S. aureus-caused staphylococcal infections. The significant interacting amino acids are Ala61, Arg84, Ile86, Ile175, Ile51, Asn54, Asp53, Ile56, Asp57, Glu58, Ser55, Ile102, Val131, Val99, Leu138, Ser129, Leu103, and Glu50 residues with a binding energy of −8.3 kcal/mol when novobiocin is docked against the DNA gyrB gene as shown in Figure 6. Novobiocin was selected as the reference compound for our investigation to find a treatment employing bioactive components generated from naturally produced plant metabolites since it is a synthetic derivative and to determine the alternate route for a naturally derived source.
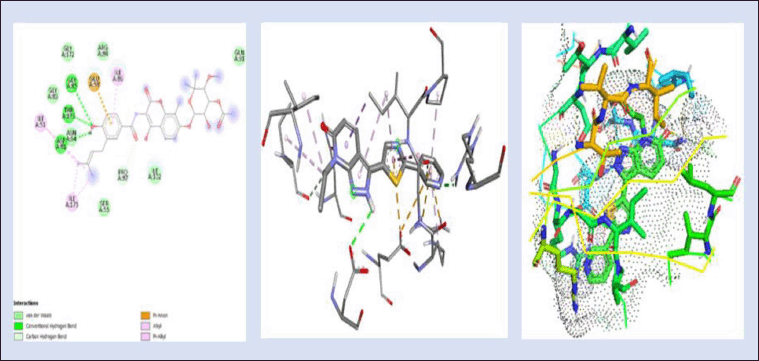 | Figure 6. 2D and 3D interaction of novobiocin (reference ligand) with DNA gyrB gene (−8.3kcal/mol) and 3D visualization of the binding pocket (dot surface view). [Click here to view] |
Using Autodock, molecular docking was used to anticipate the interaction between the binding site of a target receptor and the target ligand. It determines which orientations of a ligand within a protein’s active site are most energetically advantageous by using sampling algorithms and scoring systems. The goal of this specific active site, which was derived via complex docking and visualized utilizing Discovery Studio, is to comprehend how ligands adhere to proteins, which aids in lead optimization and drug creation.
Figure 7 shows the 2D and 3D interaction of lead 1 with DNA gyrB gene with a binding energy of −6.23 kcal/mol, which is lesser than the binding energy of novobiocin, since it is a plant-based derived compound. The interactions between the protein and the ligand are found to be Asp81, Ser55, Glu58, Asn54, Thr173, Ile51, Val79, Ile175, Leu103, Ile86, Ile102, and Gly85. Out of these 12 residues, 7 residues are nonpolar amino acids, namely, Ile, Val, Leu, and Glu are involved in this interaction, which aid in improved binding toward the protein and the ligand.
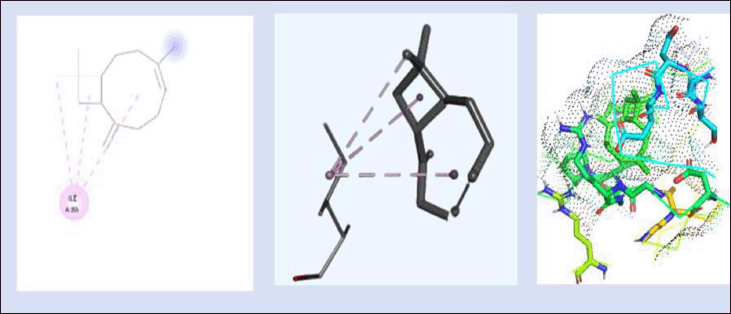 | Figure 7. 2D and 3D interaction of lead 1 (Bicyclo [7.2.0] Undec-4-Ene,4,11,11-Trimethyl-8-Methylene-, [1r-(1r*,4z,9s*)]-) with DNA gyrB gene. (−6.23kcal/mol) and 3D visualization of the binding pocket (dot surface view). [Click here to view] |
Figure 8 depicts the interaction between (4-(2,2,6trimethyl-bicyclo [4.1.0] hept-1-yl)-butan-2-one) with DNA gyrB protein and the binding energy was found to be −5.70 kcal/mol. The 2D and 3D interactions are shown in Figure 8. The protein interacted with the ligand with the amino acids’ residues Gly85, Gly83, Gly172, Glu58, Ser55, Thr173, Asp81, Ile51, Asn54, Ile175, Ile102, Arg84, Ile86, and Pro87. Among these, Gly, Thr, Ile, Arg, and Pro are present in 10 out of 14 residues, which helps in the effective binding of ligand to the protein.
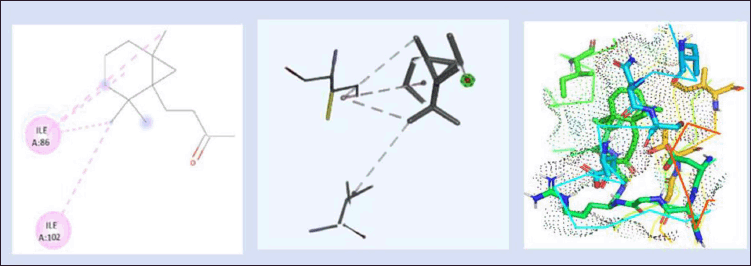 | Figure 8. 2D and 3D interaction of lead 2 (4-(2,2,6- Trimethyl-Bicyclo [4.1.0] Hept-1-Yl)-Butan-2-One) with DNA gyrB gene. (−5.70kcal/mol) and 3D visualization of the binding pocket (dot surface view). [Click here to view] |
The docking and interactions (2D&3D) of the compound Lead 3 with the protein were docked and the binding energy was found to be −6.01 kcal/mol as represented in Figure 9. According to our findings, the active sites can be quite versatile in their use of different protein domain positions and highly varied in their conservation of amino acids. Since one of the primary biochemical roles of proteins is ligand binding, identifying ligands and their binding sites is the first step toward establishing a protein’s function. Important locations, such as those directly connected to protein function, are frequently linked to amino acid sequence conservation.
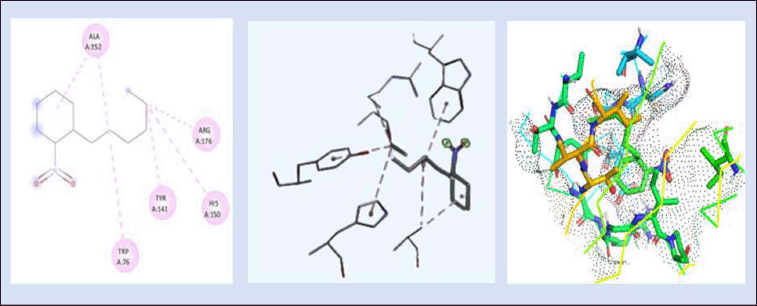 | Figure 9. 2D and 3D interaction of lead 3 (1-Hexyl-2-Nitrocyclohexane) with DNA gyrB gene. (−6.01kcal/mol) and 3Dvisualization of the binding pocket (dot surface view). [Click here to view] |
When novobiocin is docked against the DNA gyrB protein as shown in Figure 6, the amino acid residues are found to be Ala61, Arg84, Ile86, Ile175, Ile51, Asn54, Asp53, Ile56, Asp57, Glu58, Ser55, Ile102, Val131, Val99, Leu138, Ser129, Leu103, and Glu50 with the binding energy of −8.3kcal/mol. When 7-propyl-3-[2-(pyridin-3-yl)-1,3-thiazol-5-yl]-1,7-dihydro-6h-pyrazolo[3,4-b] pyridin-6-one (Reference ligand) was docked against DNA GyrB protein, the ligand interacted at the protein residues Asn54, Glu58, Ser55, Thr173, Gly85, Arg84, Ile86, Pro87, Asp81, and Arg144, as depicted in Figure 7. The binding energy of the complex was found to be −6.33 kcal/mol. The nonpolaramino acids surrounding the active site of the protein help in effective binding toward the protein. Though the complex has nonpolar interactions, this chemical complex has the lowest binding affinity among the compounds that were taken into consideration for docking. When Lead 2 is docked with the gyrase protein, the binding energy was found to be −5.70 kcal/mol. The protein interacted with the ligand at the following amino acid residues Gly85, Gly83, Gly172, Glu58, Ser55, Thr173, Asp81, Ile51, Asn54, Ile175, Ile102, Arg84, Ile86, and Pro87. Among these, Gly, Thr, Ile, Arg, and Pro are present in 10 out of 14 residues, which aid in improved binding.
When docking of the compound Lead 3 with DNA gyrB protein with the binding energy of −6.01kcal/mol as represented in Figure 9, these lead compounds have higher binding energy when docked against the DNA gyrB protein, which can significantly inhibit the protein in the Staphylococcal infection out of these seven leads, and lead 1 has 12 active sites of amino residues; out of these 12, 7 amino acids (Ile51, Val79, Ile175, Leu103, Ile86, Ile102, and Gly85) are nonpolar, which increases the binding stability of the complex.
Upon closer examination, lead 1 has the highest stacking interaction with the protein compared to all the other chemicals studied, proving that this compound is more effective against Staphylococcal infection than the chemical-based drugs. This study includes seven leads for the docking studies, and out of that lead 1 is chosen for the simulation process based on the binding affinity, protein–ligand interaction, and highest binding energy. The reference compound is taken from the PDB, which is shown as the reference compound for the protein. Thus, the other compounds that showed less binding energy compared to lead 1 are not further processed for the simulation studies.
Protein–ligand interaction profiler
Protein–ligand interaction of the complex of DNA GyrB protein with reference, lead 1, lead 2, and lead 3 compounds are demonstrated using PLIP software, and the types of ligand interaction, bond length, and distance are listed. The diagrammatic representation, including Figures 10–13, is the protein–ligand interaction of the docked protein and ligand complex. The docked complex of protein DNA GyrB with the ligand novobiocin has hydrophobic interaction, hydrogen bond, and π-cation interactions. The bond length and amino acid interactions are shown in Table 11. The docked complex of protein DNA GyrB with the ligand lead 1 has hydrophobic interaction, and the bond length and amino acid interactions are shown in Table 12. The docked complex of protein DNA GyrB with the ligand lead 1 has hydrophobic interaction. The bond length and amino acid interactions are shown in Table 13. The docked complex of protein DNA GyrB with the ligand lead 1 has a hydrogen bond. The bond length and amino acid interactions are shown in Table 14.
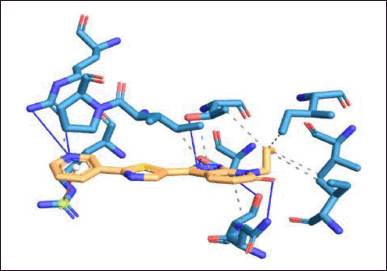 | Figure 10. Protein–ligand interaction of DNA GyrB with reference ligand. [Click here to view] |
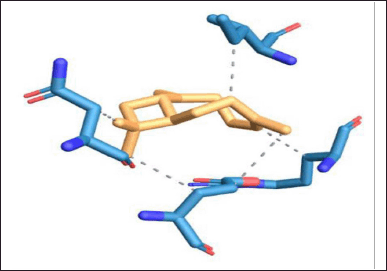 | Figure 11. Protein–ligand interaction of DNA GyrB with lead 1 ligand. [Click here to view] |
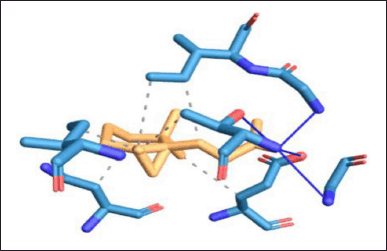 | Figure 12. Protein–ligand interaction of DNA GyrB with lead 2 ligand. [Click here to view] |
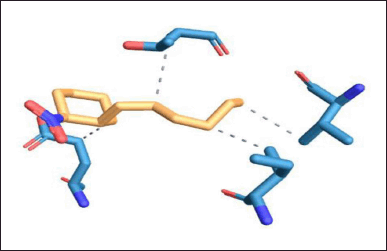 | Figure 13. Protein–ligand interaction of DNA GyrB with lead 3 ligand. [Click here to view] |
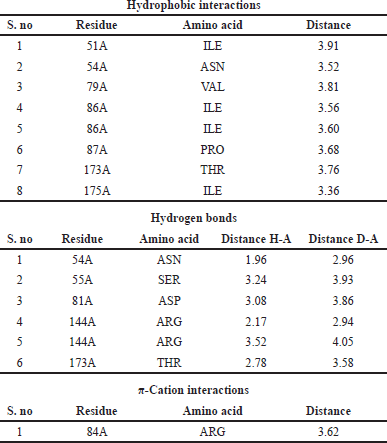 | Table 11. Interaction between ligands and amino acid residues with bond length of the DNA GyrB with reference. [Click here to view] |
 | Table 12. Interaction between ligands and amino acid residues with bond length of the DNA GyrB with lead 1. [Click here to view] |
 | Table 13. Interaction between ligands and amino acid residues with bond length of the DNA GyrB with lead 2. [Click here to view] |
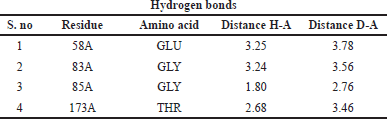 | Table 14. Interaction between ligands and amino acid residues with bond length of the DNA GyrB with lead 3. [Click here to view] |
Class and biological properties of the lead compounds
Table 15 depicts the class and biological properties of lead compounds. Most of the lead compounds show antibacterial activity, anti-inflammatory activity, and analgesic properties.
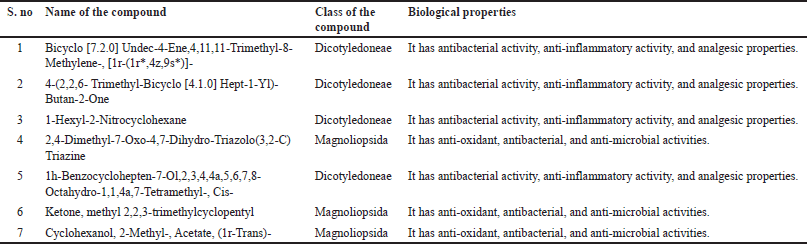 | Table 15. Class and biological properties of lead compounds. [Click here to view] |
Pharmacokinetic properties in silico analysis
Table 16 describes the virtual screening of phytochemicals using the Swiss-ADME platform that follows Lipinski’s rule of five. The seven phytochemicals lead 1, lead 2, lead 3, lead 4, lead 5, lead 6, and lead 7, which followed Lipinski’s rule of 5, are further laid open to molecular docking against the protein (DNA gyrB).
 | Table 16. Virtual screening of phytochemicals using Swiss-ADME. [Click here to view] |
Pharmacokinetic prediction
The pharmacokinetic characteristics of putative bioactive substances were predicted, researched, and recorded in Tables 17 and 18. Every compound breached the blood–brain barrier (BBB) except the novobiocin.
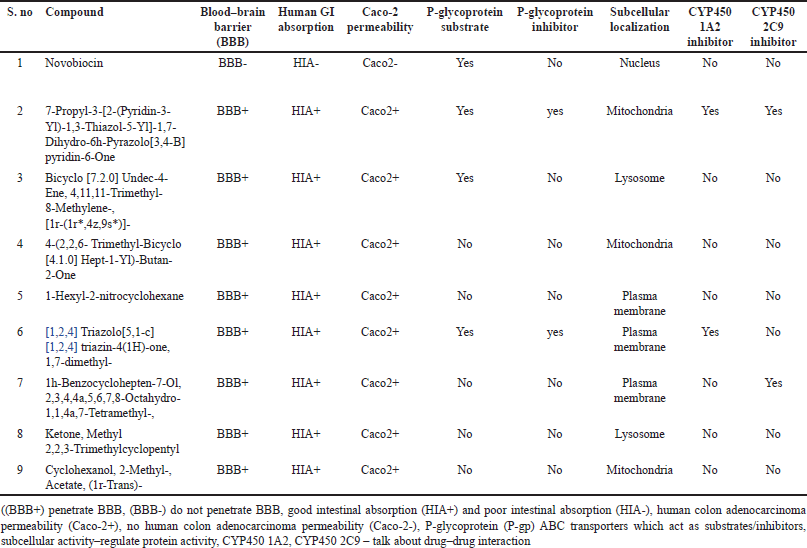 | Table 17. Pharmacokinetic properties of phytochemicals. [Click here to view] |
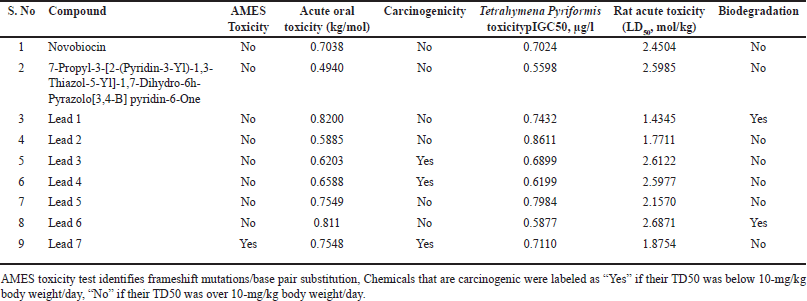 | Table 18. Toxicity properties of phytochemicals. [Click here to view] |
BBB indicates whether the compound can cross the blood–brain barrier. “BBB+” denotes that the compound can cross the blood–brain barrier, while “BBB-” indicates that it cannot. Human GI absorption (HIA) indicates whether the compound is absorbed in the human gastrointestinal (GI) tract. “HIA+” means that the compound is absorbed, while “HIA-” means that it is not. Caco-2 permeability indicates the permeability of the compound through Caco-2 cell monolayers, which are commonly used to study intestinal absorption and drug transport. “Caco2+” means that the compound is permeable, while “Caco2-” means that it is not. P-glycoprotein substrate indicates whether the compound is a substrate for P-glycoprotein, a drug efflux transporter involved in multidrug resistance. “Yes” means that it is a substrate, while “No” means that it is not. P-glycoprotein inhibitor indicates whether the compound inhibits P-glycoprotein activity. “Yes” means that it inhibits, while “No” means that it does not. Subcellular localization indicates the location within the cell where the compound is localized. Examples include nucleus, mitochondria, lysosome, and plasma membrane. CYP450 1A2 inhibitor indicates whether the compound inhibits the activity of cytochrome P450 1A2 enzyme. “Yes” means that it inhibits, while “No” means that it does not. CYP450 2C9 inhibitor indicates whether the compound inhibits the activity of cytochrome P450 2C9 enzyme. “Yes” means that it inhibits, while “No” means that it does not.
AMES toxicity indicates whether the compound shows toxicity in the AMES test, which is a bacterial reverse mutation assay used to assess the mutagenic potential of chemicals. “Yes” indicates toxicity, while “No” indicates no toxicity. Acute oral toxicity represents the acute toxicity of the compound when administered orally. It is measured in LD50, which is the lethal dose required to kill 50% of the test population. It is expressed in mol/kg. Carcinogenicity indicates whether the compound is carcinogenic. “Yes” indicates that it is carcinogenic, while “No” indicates that it is not. Tetrahymena pyriformis toxicity (pIGC50, μg/l) is a type of ciliated protozoan often used as a model organism in toxicity testing. This column provides the toxicity of the compound against t. pyriformis, expressed as pIGC50 (half maximal inhibitory concentration) in micrograms per liter (μg/l). Rat acute toxicity (LD50, mol/kg) represents the acute toxicity of the compound when administered to rats. It is measured in LD50, which is the lethal dose required to kill 50% of the test population. It is expressed in mol/kg. Biodegradation indicates whether the compound undergoes biodegradation, meaning that it can be broken down by biological processes into simpler compounds. “Yes” indicates that it undergoes biodegradation, while “No” indicates that it does not. All the compounds are noncarcinogenicity except lead 3, lead 4, and lead 7.
Molecular dynamics
The firmness and dynamics of the free protein (PDB ID: 5D7D) were modeled for 100 ns using a ligand with high affinity and low binding energy to examine the RMSD and RMSF. The stability of protein increases with an increase in RMSF and decreases in RMSD. Around one lakh distinct frames were generated using the simulation study for the single protein–ligand complex. The molecular structure at that precise moment was captured in a single frame. The target ligand, lead 1, and the reference ligand (novobiocin) fluctuations against the target protein (DNA gyrB) are depicted in Figures 14 and 15.
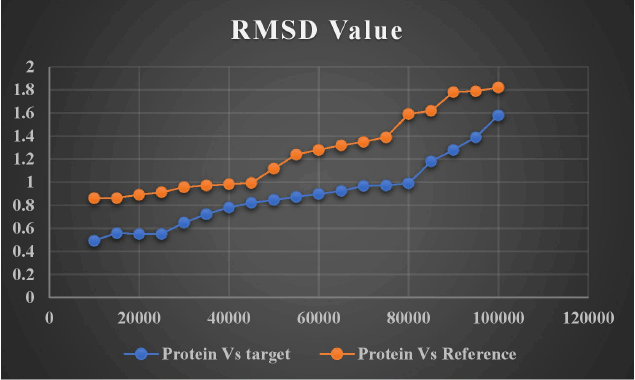 | Figure 14. RMSD of target and reference ligand. [Click here to view] |
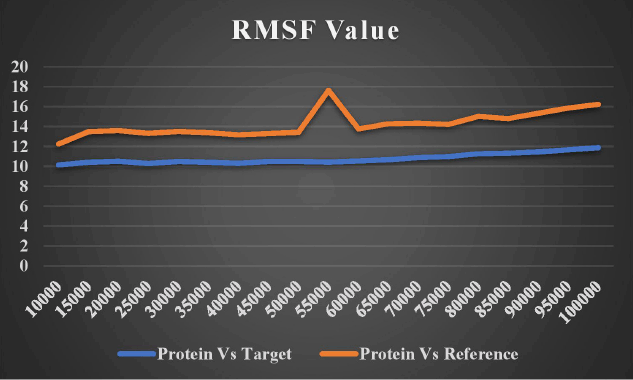 | Figure 15. RMSF fluctuation of target and reference ligand. [Click here to view] |
Hydrogen bonding is one of the most important interactions for maintaining the stability of the protein–ligand complex. Figure 16 shows how DNA gyrB bound to lead 1 and the reference chemical (novobiocin) showed a higher number of average hydrogen bonds, indicating the efficacy of these molecules as DNA gyrB inhibitors. As the reference molecule (novobiocin) is a commercially available prescription chemical medication that is used to treat patients with staphylococcal infection, a bioactive ingredient lead 1 can be a helpful herbal formulation substitute.
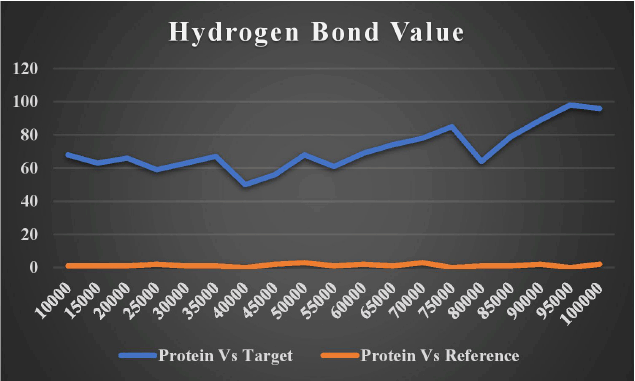 | Figure 16. Number of hydrogen bond interactions of target and reference ligand. [Click here to view] |
Figure 17 compares the target protein (DNA gyrB) to the solvent-accessible surface area (SASA) of the target ligand lead 1 (Bicyclo [7.2.0]Undec-4-Ene,4,11,11-Trimethyl-8-Methylene-,[1r-(1r*,4z,9s*)]-) and co-crystal reference ligand (7-Propyl-3-[2-(Pyridin-3-Yl)-1,3-Thiazol-5-Yl]-1,7-Dihydro-6h-Pyrazolo[3,4-B]pyridin-6-One). The region of a protein that is sufficiently exposed to contact with nearby solvent molecules is referred to as SASA. SASA is a deciding factor in studies on protein stability and folding. The more points we receive, the more of the molecule is sticking out into the water. Low scores imply that the molecule is more thoroughly incorporated into the protein. Lower scores are given to the target protein (DNA gyrB) compared to the target ligand lead 1. The ideal molecule to investigate should typically be the one with the lowest score for therapeutic applications.
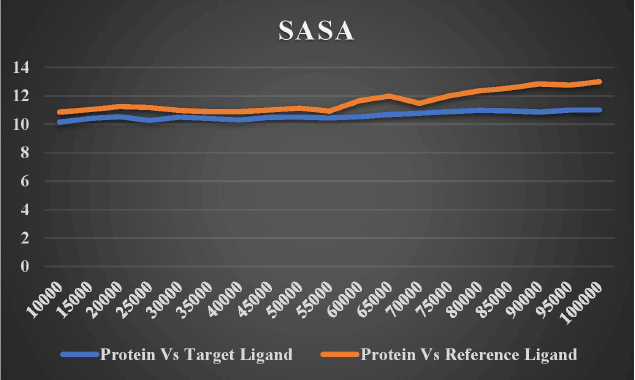 | Figure 17. Solvent accessible surface area (SASA) of both target ligand and reference ligand versus target protein. [Click here to view] |
The dynamic behavior of the screened compounds is examined for low energy profiles employing MM/PBSA, which is utilized for docked structure postprocessing in addition to the dependability of inside the flexible binding pocket, complex binding. The lead molecule and the standard drug appear to be fit precisely in the binding site and have a low energy profile, based on the 100-ns protein–ligand complex simulation and the MM-PBSA binding free energy. The literature that is currently available indicates that the compounds in MM/PBSA with lower energy profiles would make good candidates for additional experimental investigation. Table 19 and Figures 18 and 19 show the MM/PBSA calculation of reference and lead 1 ligand.
 | Table 19. MMPBSA energy calculation of reference and target ligand. [Click here to view] |
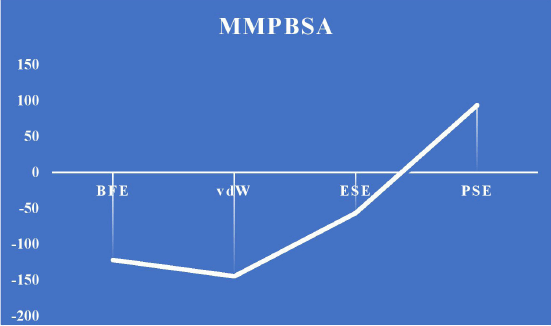 | Figure 18. MMPBSA energy calculation for lead 1. [Click here to view] |
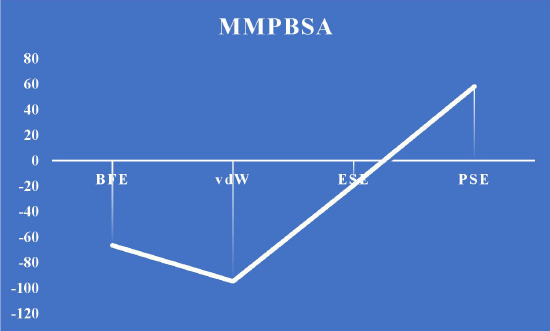 | Figure 19. MMPBSA energy calculation for reference. [Click here to view] |
This research focuses on the discovery of new plant-based drug to treat staphylococcal infection, which seems like a promising endeavor. Using molecular simulation studies, system biological tools and docking analysis proves that the lead 1 (Bicyclo [7.2.0] Undec-4-Ene,4,11,11-Trimethyl-8-Methylene-, [1r-(1r*,4z,9s*)]-) being extracted from the traditional medicinal plants may be used as novel therapeutic target lead toward the staphylococcal infections.
CONCLUSION
The use of traditional herbal remedies has been asserted and subsequently validated by humans through research investigations and trial-and-error experiments. Through network analysis, the DNA GyrB gene was exposed to docking and MD modeling for 100 ns using the plant-derived ligand firmness, a putative bioactive chemical with high binding energy (−6.23 kcal/mol), using the Autodock and NAMD tools. In addition to improving our knowledge of molecular pathways, the research reported here suggests lead 1 (Bicyclo [7.2.0] Undec-4-Ene,4,11,11-Trimethyl-8-Methylene-, [1r-(1r*,4z,9s*)]-) as a potential target for therapy. The development of medicines for staphylococcal infections will benefit from these results, which will in the not-too-distant future lead to a greater understanding of the molecular and structural mechanisms behind the action of these medications. The tremendous developments in the areas of computational research and in silico techniques can offer very significant advantages for the regulatory standards and the safety profile assessment used by the pharmaceutical sector.
ACKNOWLEDGMENT
The authors are thankful to the management of Vels Institute of Science, Technology, and Advanced Studies (VISTAS), Chennai, Tamil Nadu, India, for providing all the facilities to conduct this research work. They also thank Dr. Priyanka Purkayastha for helping in the simulation analysis.
AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICS APPROVALS
This study does not involve experiments on animals or human subjects.
AVAILABILITY OF DATA AND MATERIALS
This research article includes all the data that were obtained and studied.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000 Jan 1;13(1):16–34.
2. Blanche F, Cameron B, Bernard FX, Maton L, Manse B, Ferrero L, et al. Differential behaviors of Staphylococcus aureus and Escherichia coli type II DNA topoisomerases. Antimicrob Agents Chemother. 1996 Dec;40(12):2714–20.
3. Guo Y, Song G, Sun M, Wang J, Wang Y. Prevalence and therapies of antibiotic-resistance in Staphylococcus aureus. Front Cell Infect Microbiol. 2020 Mar 17;10:107. doi: CrossRef
4. Foster TJ. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol Rev. 2017 May 1;41(3):430–49.
5. Menzel R, Gellert M. Regulation of the genes for E. coli DNA gyrase: homeostatic control of DNA supercoiling. Cell. 1983 Aug;34(1):105–13. doi: CrossRef
6. Rajakumari K, Parthiban BD, Rishikesan S. Phytochemical analysis and antibacterial activity of traditional plants for the inhibition of DNA gyrase. Biomedicine. 2023 Sep 18;43(4):1203–8.
7. Balandrin MF, Kinghorn AD, Farnsworth NR. Plant-derived natural products in drug discovery and development: an overview. Washington, DC: ACS Publications; 1993. 2–12 pp. doi: CrossRef
8. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. “STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets.” Nucleic Acids Res. 2019;47(D1):D607–13. doi: CrossRef
9. Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. “Cytoscape automation: empowering workflow-based network analysis.” Genome Biol. 2019;20:1–5. doi: CrossRef
10. Nath R, Rajakumari K, Thiruchelvi R, Romauld S, Vivek P. Chitosan nanoparticle encapsulated with Terminalia chebula – in vitro antidiabetic activity. Oxid Commun. 2023; 46(1):139–50.
11. Shoichet BK, McGovern SL, Wei B, Irwin JJ. “Lead discovery using molecular docking.” Curr Opin Chem Biol. 2002;6:436–49.
12. Sousa M, Pozniak A, Boffito M. “Pharmacokinetics and pharmacodynamics of drug interactions involving rifampicin, rifabutin and antimalarial drugs.” J Antimicrob Chemother. 2008;62(5):872–8. doi: CrossRef
13. Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018 Sep 19;99(6):1129–43. doi: CrossRef
14. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. “Scalable molecular dynamics with NAMD.” J Comput Chem. 2005;26(16):1781–802. doi: CrossRef
15. Venkatachalam K. Vitex negundo: medicinal values, biological activities, Toxicity studies and Phytopharmacological actions. Int J Pharm Phytopharmacol Res. 2012;2(2):126–33.