
INTRODUCTION
Quality by design (QbD) is a strategic, new, and optimistic approach to developing any pharmaceutical product. However, more evidence is needed for the use of QbD for the development of analytical methods at the industrial scale. QbD is an emerging tool for improving product quality at each step of pharmaceutical product development. In particular, QbD can help reduce project costs at the pilot scale while minimizing chemical costs, human resources, and time [1,2]. The proposed study aims to develop a hybrid analytical method that can overcome this existing gap at the industrial scale by using the QbD approach. Quality checks play a pivotal role in pharmaceutical product development. There are various pharmaceutical companies that manufacture antidiabetic drugs, but unfortunately, most of them use conventional methods for quality control checks. At the same time, the emerging regulatory market requires quality products with low cost, reliability, and customer satisfaction. The QbD approach can help in mitigating the above obstacles. Regulatory agencies such as the International Council of Harmonization (ICH) and the US Food and Drug Administration have implemented guidelines for pharmaceutical product development [3]. By using the QbD approach, the quality problem can be detected early, and the overall product cost can be minimized by the manufacturer. In the modern era, quality competition is crucial to achieve quality products and capture a market share with massive shifting. In this study, glimepiride (GLP) was used as a model drug. It is an antidiabetic drug that is used for the therapeutic management of type II diabetes. This research reports that QbD-based analytical method optimization requires less time and has a reduced chemical cost [4–9].
Analytical methods are critical elements in product development due to their roles in process development and product quality control. Inaccurate analytical methods can cause results that are not reliable, potentially providing misleading information that could harm the drug development program. In an endeavor to address such plausible crucial issues, different pharmaceutical regulatory agencies, such as the ICH and US FDA, have adopted QbD principles to circumvent these quality crises [10]. Recently, ICH has announced a new guideline, ICH Q14, on analytical procedure development and revision regarding drug products and drug substances [11]. The proposed research aims to develop a simple, cost-effective, rapid, and sensitive RP-HPLC method using the QbD approach. Three-level factorial designs were employed for the experimental design: flow rate (X1), pH of the mobile phase (X2), and column oven temperature (X3). At the same time, the peak area (Y1), peak height (Y2), and number of theoretical plates (NTPs) (Y3) were used as responses. The experimental design was validated by statistical analysis using ANOVA. The analytical method was validated as per the ICH Q2 (R1) guidelines with parameters such as system suitability, method precision (interday and intraday), recovery, linearity, LOD, LOQ, accuracy, solution stability, robustness, and ruggedness. Furthermore, the developed method was transferred from the originator to the receiver laboratory as per US Pharmacopeia (USP) <1224>. In this way, the RP-HPLC method for detecting GLP from pharmaceutical drug products (transdermal patches) was developed and validated. The same method was employed for bioanalytical application. However, we partially validated the method in mouse plasma. GLP was extracted from mouse plasma via a liquid?liquid extraction technique and injected via HPLC for quantification. GLP is a Biopharmaceutical Classification II (BCS-II) drug that has low solubility and high permeability. Based on the polarity of the compound, the RP-HPLC method was selected for quantifying GLP.
MATERIALS AND METHODS
Chemicals and reagents
GLP was procured from Yarrow Chem Products, Mumbai, India. Chromatographic solvents such as acetonitrile (#SE0SF70584), methanol (#SC7SF67277), orthophosphoric acid (Emplura® #1.93403.0521), and ammonium acetate (#61855405001730) were obtained from Merck India. Ammonium hydroxide solution (#A669500) and formic acid (#2173388) were obtained from Thermo Fisher Scientific. Syringe filters, such as Axiva PVDF, Sterile, 0.22 µM (#SFNY25 RB), Nylon Randisc, sterile 0.22 µM (#RANKNY4513SF-100PB), and Sartorius Sterile, 0.22 µM (#20232103), were used in this study. Syringes (with volumes of 5 ml and 10 ml) were procured from Sigma. An HPLC column was procured from Phenomenex (Sr. No # 5701–0059). All the solvents and chemicals used for the study were of chromatographic grade.
Instrumentation
A Shimadzu binary HPLC system (SIL-20AC HT), a photodiode array detector (PDA) model (#SPD-M20A), a binary pump model (#LC20DA), a column oven model (#CT0-10AS VP) and a mobile phase filtration assembly (Pall Corporation, model #NR047100), an autoinjector with a loop volume of up to 100 µl, and a single degasser unit connected to the mobile phase were used for method development and validation. In the wet laboratory, we used various analytical instruments and equipment for sample preparation, such as an analytical balance (Make: Metter Toledo Model No. ME204/A04), a vortex shaker (Make: IKA, Model # VG 3 S22), a probe sonicator (RK 103H, BANDELIN Sonorex), a pH meter (Make: Thromo Scientific), a centrifuge (Make: Eppendorf Model: #5430-R), a nitrogen concentrator, a calibrated volumetric flask (Volume 10 ml, 25 ml and 50 ml), and a syringe filter. Furthermore, the method was transferred to the receiver laboratory at the Shimadzu HPLC system model (#LC2010C HT) equipped with a PDA detector (Model #SPD-M20A3).
Chromatographic conditions
The method was developed using a Shimadzu HPLC system with a PDA detector on a Thermo Fisher Scientific HPLC column (C18, 150 × 4.6 mm), a particle size of 4 µm, an injection volume of 15 µl, a column oven temperature of 40°C, and a sample cooler temperature of 15°C with a run time of 5 minutes. The isocratic flow rate was programmed using mobile phase A (50 mM ammonium acetate buffer pH 4.0 with 0.1% formic acid) and mobile phase B (acetonitrile). A flow rate of 0.88 ml/minute with a gradient ratio of 40:60 was used. GLP was detected at a wavelength of 228 ± 2 nm.
Method Development
A cost-efficient method was developed to meet the industrial requirements of both analytical and bioanalytical samples. Both methods were validated as per the ICH Q2 (R1) guidelines [12]. The optimized mobile phase and buffer solution were used to carry out the study. A low-cost solvent was used for the quantification of GLP from transdermal patches and rat plasma. An HPLC detector was selected based on the present chromophore group in the chemical structure of GLP (Fig. 1). Similarly, the dilution, acetonitrile: methanol ratio (50:50), C18 column, and final run time were determined based on the separation of the compounds from the column and the system suitability, as described in USP chapter 621. Initially, 100 µg/ml GLP was prepared in diluent media and scanned into a PDA detector from 200 to 800 nm.
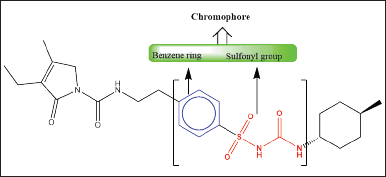 | Figure 1. The chemical structure of GLP contains two chromophore groups, highlighted in red as sulfonyl groups and highlighted in blue as benzene groups. Both functional groups are responsible for absorbing UV light at different wavelengths. The electron transitions involved are the (C=O, n → π* and π→π*) transitions. [Click here to view] |
Method optimization using QbD
The developed analytical method was optimized through the QbD approach using the Box?Behnken design for the optimization of the mobile phase, column oven temperature, and flow rate. A three-level factorial design was chosen for the final optimization of the dependent factors (X1) mobile phase pH, (X2) flow rate, and (X3) column oven temperature. The experiments were designed at three center points with three response factors, namely, peak area (Y1), HPLC height (Y2), and United States Pharmacopeia number of theoretical plates (USP NTP) (Y3). A total of 18 runs were suggested by the software. Thereafter, each suggested run was injected into the HPLC system, and the response factors were determined individually. Subsequently, all the response factors were incorporated into the QbD application and analyzed using quadratic equation 1. Ideally, the model should be significant, and the lack of fit should be nonsignificant. The quadratic equation was used to determine the final method. The factor’s criteria were set for A, B, and C in the range A–0.8 to 1.2 ml/minute, B–4.00 to 5.00 pH, and C–40.00°C–45.00°C. Eventually, the software predicted an optimistic run with a desirability value closer to 1 [2,13].
ax2 + bx + c = 0 (1)
Method validation
System suitability
The system suitability test (SST) is the initial parameter for HPLC-based analytical method development. By evaluating this parameter, a standard stock solution of GLP was prepared at a concentration of 100 µg/ml in the diluent. The same procedure was followed for mouse plasma except for sample preparation. The SST sample was prepared from a mouse plasma stock sample at a concentration of 100 µg/ml. Furthermore, both SST samples were injected into the RP-HPLC system (n = 6 injections), and the system suitability parameters, including the tailing factor, percentage relative standard deviation (% RSD), NTP, and retention time (RT), were verified. The following acceptance criteria should be met as per the United States Pharmacopeia chapter 621 (USP-621): NTP not less than 2000, tailing factor not more than 2, %RSD not more than 2%, and RT ± 10% of the principal peak [14].
Preparation of standard stock solution in diluent media
GLP (50.02 mg) was weighed and transferred to a 50 ml volumetric flask containing 20 ml of diluent. This flask was vortexed for 5 minutes, followed by sonication for 10 minutes until the particles completely dissolved, after which the volume was adjusted to the mark with the same diluent. The concentration of the final stock solution was 1 mg/ml.
Collection of mouse plasma
Mouse plasma was collected from the BITS-PILANI Hyderabad animal facility with ethical approval. No BITS-HYD/IAEC/2020-31. We declare that the experimental procedures and animals were taken care of as per the Animal Research: reporting of in vivo experiments (ARRIVE) guidelines.
Preparation of standard stocks in mouse plasma
The stock solution was prepared according to the method reported by Priyanka Maurya et al. [15]. GLP (2.01 mg) was weighed and transferred to a 2 ml sterile tube containing 1 ml of mouse plasma. The tube was vortexed for 5 minutes and kept on the benchtop for 10 minutes to absorb the drug in mouse plasma. The liquid?liquid extraction technique was used for the extraction of GLP from mouse plasma, and 1 ml of diluent media (acetonitrile: methanol,1:1) was added for protein precipitation, followed by centrifugation at 15,000 rpm at 5°C for 20 minutes. The supernatant was collected in another 2 ml sterile tube, and a 1,000 µg/ml dilution was made and filtered with a 0.22 µm nylon syringe filter [16].
Sensitivity (LOD and LOQ)
The LOD and LOQ are used for method validation to determine the detection and quantification range of the analyte. ICH Q2 (R1) was excluded from the method validation. Herein, bioanalytical method development and validation were carried out on mouse plasma. The previous dilution in the linearity section was increased to 0.024 µg/ml. A total of 7 dilutions (1.562, 0.781, 0.390, 0.195, 0.097, 0.048, and 0.024 µg/ml) were considered for the estimation of the LOD and LOQ. It was calculated based on a recently reported method by Marie et al. [10] (signal-to-noise ratio).
Matrix effect
The plasma matrix effect was established using the postextraction method. We evaluated the matrix effect by comparing the linearity slope in both media, e.g., plain diluent and blood plasma. The final matrix effect was calculated using equation 2.
Linearity and range
Serial dilutions were made from the respective standard stock solutions (at a concentration of 1 mg/ml) to establish the linearity and range of the analytical procedure. The range was established with lower and upper concentrations at three levels with accuracy, precision, and linearity. A serial dilution was made from the respective stock solution (100 µg/ml to 3.125 µg/ml). A total of 6 different concentrations were prepared and injected into the HPLC system. The same protocols were used for mouse plasma, and the linearity and range of GLP were established. The correlation coefficient was calculated. R2 should not be greater or less than 0.999 ± 0.004.
Specificity
This approach is a part of the analytical method for validating drug products. The purity angle should be less than the purity threshold [17]. Three different test samples were prepared: a transdermal patch, a pure drug (glimepiride), and a placebo at a concentration of 100 µg/mL. Subsequently, the samples were injected into the HPLC system, and the percent interference was calculated. The percent interference should not be more than 0.2% as per USP limits.
Method precision
Precision was assessed through both interday and intraday evaluations in the same laboratory using the same instrument. Six precision samples were prepared to establish the precision and reproducibility of the method. A sample concentration of 100 µg/ml was prepared using the same diluent media for the transdermal patch. The same concentration was used for the preparation of the standard sample, and both of these samples were injected into the HPLC system. Similarly, the determination of method precision in mouse plasma was performed except for intraday precision. Eventually, the % RSD (n = 6) was calculated, and it should not be more than 2% [18].
Accuracy
The accuracy ensures the closeness of the agreement with the accepted reference values. This means that the obtained results should be accurate, precise, and authentic. The experiment evaluated three different concentrations, 150 µg/ml, 100 µg/ml, and 50 µg/ml, on the same day. Each set of concentrations was injected into the HPLC system (n = 3 injections). Accurate samples were prepared in diluent media and mouse plasma. The final dilution was made in diluent media for the mouse plasma sample. The experimental data were statistically analyzed, and the % was calculated as per equation 3 accuracy, ± SD. The results were confirmed at a 95% confidence interval. The recovery should be 100% ± 2% as per the 21-code of federal regulations (21-CFR) 211.194 [(a) (2)] and the United States Pharmacopeia-National Formulary (USP-NP) [19,20].
Robustness
This analytical procedure is designed to withstand small method changes and deliberate parameter variations. While this parameter can be omitted based on QbD analysis, we followed the ICH Q2 (R1) guidelines and validated it with slight method adjustments, including ±10% flow rate, ±10% column oven temperature (°C), ±10% mobile phase composition, ±10% mobile phase pH, and ± 2 nm wavelength changes. All the experimental results were analyzed through HPLC, and the % accuracy was calculated by equation 3. These variations should not be strongly impacted by NTP, tailing factor, RT, or percent accuracy [21].
Filter paper interference
Filter paper interference was validated by using different syringe filters, i.e., 0.22 µm nylon syringe filter, 0.22 µm Whatman PVDF filter paper, and 0.22 µm sterile Alxie filter. A sample concentration of 100 µg/ml was prepared, and the sample was filtered through a syringe filter and further injected into the HPLC system. Finally, the percent accuracy was calculated using equation 3. The recovery of each filter ranged from 95.0% to 105.0%.
Solution stability
Solution stability is not an integral part of analytical method validation as per ICH Q2 (R1). However, pharmaceutical companies must work with routine samples. In this case, solution stability was established for up to 24 hours at different time intervals and under two different storage conditions, i.e., room temperature and refrigeration. Thus, a sample with a concentration of 25 µg/ml was prepared and kept at room temperature (25°C ± 2°C) in a refrigerator (8°C ± 2°C). The % absolute difference was calculated using equation 4 by injecting each interval and fresh sample at the same concentration.
Technology transfer
Technology transfer is a prerequisite before routine analysis. When the validation laboratory is geographically distant from the pharmaceutical manufacturing unit, analytical technology transfer (AAT) is necessary. This process involves a documented transfer of validated methods from the originator to the receiver laboratory [22]. In this instance, the AAT procedure was demonstrated from the originator lab “GLP laboratory-A” to the receiver “laboratory-B”. The experiments were conducted by two expert analysts at both sites in accordance with USP-NP chapter 1224 [23]. Thereafter, a comparative analysis was performed with the same method, the same lot of samples, and the same test with the same acceptance criteria at both sites [24]. In this study, an intermediate precision test was carried out by injecting a 100 µg/ml precision sample (n = 6). The same experiment was repeated at the receiver site, and a comparative evaluation of the results from both laboratories was carried out. The percent RSD was not permitted to exceed 2% in either laboratory for assay method validation. The experimental data were interpreted as per USP-NP general chapter 1010 [25].
Industrial applications
The proposed validated analytical method was employed for the routine analysis of both the solid oral formulation of GLP and the transdermal patch [26]. The GLP-loaded transdermal patch was tested by cutting a 1 cm2 section, which was subsequently transferred to a 50 ml volumetric flask containing 25 ml of media. This mixture was sonicated until complete dissolution, and the remaining volume was adjusted to the mark. A suitable dilution of 100 µg/ml was prepared, and the resulting solution was filtered through a 0.22 µm syringe filter. The filtered solution was then added to HPLC vials and injected into the HPLC system. Simultaneously, a standard sample with the same concentration of 100 µg/ml was prepared and injected into the HPLC system. The HPLC area was ultimately calculated.
For the assay of GLP tablets, commercially obtained tablets were utilized. The equivalent weight of the powdered sample was transferred into a 50 ml volumetric flask containing 25 ml of media. The flask was sonicated until the active particles were fully dissolved. The subsequent steps mirrored those of the GLP-loaded transdermal patch assay.
Regarding the liposomal formulation, GLP-loaded liposomes were prepared using a film hydration method, with a final formulation containing 2 mg/ml of the drug. The quality of the liposomal formulation was assessed by extracting GLP from it using the probe-sonication method. Specifically, 1 ml of the liposomal formulation was transferred to a 5 ml Eppendorf tube, and an equal volume of media was added. The mixture underwent 30 probe cycles with probe sonication, with 5 minutes of “On” and 1 minute of “Off” intervals.
RESULTS
Method development
Based on the chromophore groups present in the GLP chemical structure (Fig. 1), a PDA detector was chosen. The peak purity and λmax of GLP were determined with a purity angle of –0.04946, a purity threshold of 1.000, and a λmax of 277 ± 2 nm (Fig. S2 G and H). Two polar solvents were initially chosen for proper analyte separation: (A) 50 mM ammonium acetate buffer containing 0.1% formic acid at pH 4.5 ± 0.5 and (B) acetonitrile. Subsequently, the isocratic run was conducted with a flow rate of 1 ± 0.2 ml/minute using a composition of 60:40, and the compound was eluted from the column at approximately 4 ± 1.2 minutes within a 10-minute run time (Fig. 2a). It is important to note that the analyte RT shifted to approximately 3.6 minutes ± 0.36% when the mobile phase composition ratio was altered to 45:55 (Fig. 2b). Subsequently, the method was optimized, and the RT was found to be 2.8 minutes with a 5 minute run time (Fig. 2c). All SSTs, including NTP, % RSD, and telling factor, were found to be in compliance with USP chapter 621 (Fig. S4).
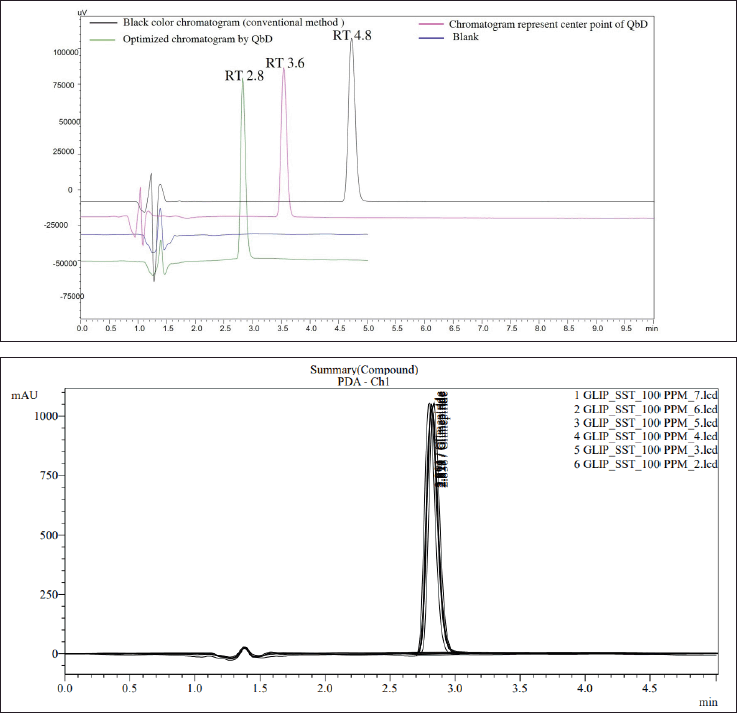 | Figure 2. In overlay chromatogram view 2A, (A) shows the chromatogram with a RT of 2.8 minutes, representing the final optimized method. (B) displays a chromatogram with RT 3.8 minutes applying QbD principles at the center point. (C) depicts a standard chromatogram with RT 4.8 minutes using a conventional HPLC method, while (D) represents the blank chromatogram. In overlay chromatogram view 2B, system suitability chromatographs of GLP at a concentration of 100 PPM are presented. All parameters meet the acceptance range specified in USP chapter 621. [Click here to view] |
Method Optimization using QbD
Next, the developed analytical method was optimized using a Box?Behnken surface design. Three factors were incorporated into the experimental design: A - HPLC area, B - HPLC height, and C - NTPs. The experiment was successfully designed at two different levels (Table S1a). Subsequently, the software predicted 18 runs, including 6 center points (Table 1). The summary design indicated a quadratic model through polynomial analysis. The applied QbD model showed significant differences, with p values of < 0.0001, 0.0011, and 0.0001 for three distinct response factors. Additionally, the R-squared (r2) values were determined to be 0.9995, 0.9267, and 0.9611, aligning with the specified limits for the respective response factors. The acceptance criteria were set as “maximum” for all three response factors, resulting in an HPLC area of 304450, an HPLC height of 135000, and an NTP-USP of 7,000. Based on these criteria, the optimal solution was determined by the QbD software, selecting a method with a desirability value of 0.864. The 3D model graphs for response factors with desirability are illustrated in Figure 4A–D. Subsequently overlay plot of optimized results are reported in Figure S1 and standard error experiment design (Box–Behnken) graph depicted in Figure S3.
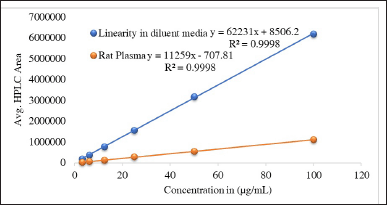 | Figure 3. The linearity of glimepiride graph was confirmed across a concentration range of 3.12 to 100 μg/ml using six different concentrations. The experiment was replicated three times (n = 3) for both the diluent media and rat plasma. Consequently, the coefficient of determination (R2) was found to be 0.999 for both sample types. [Click here to view] |
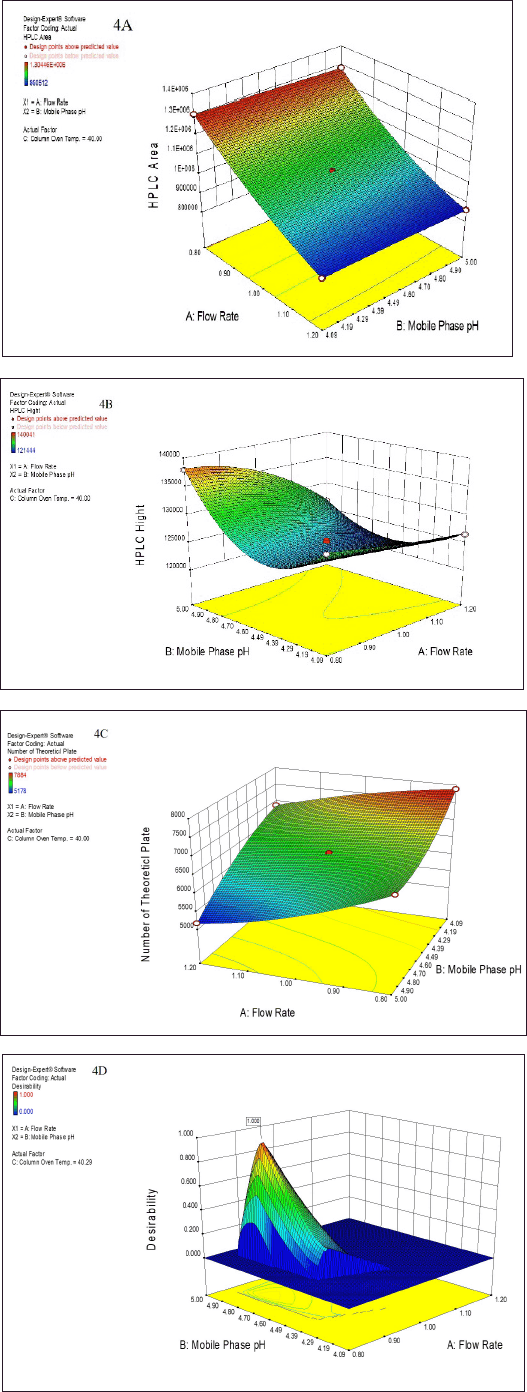 | Figure 4. The summarized surface-optimized graph presents a three dimensional (3D) model depicting three distinct response factors. These optimized graphs visually represent how variations in mobile phase pH and flow rate influence the following response factors: (A) HPLC Area, (B) HPLC height, and (C) NTP. Additionally, graph (D) represents the desirability value, which approaches 1, indicating the optimal method for HPLC analysis as highlighted above the graph. [Click here to view] |
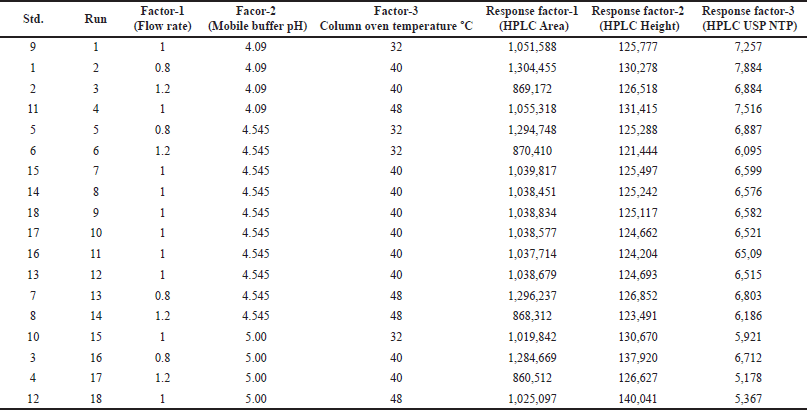 | Table 1. Design matrix as per Box-Behnken design (BBD) for optimization of flow rate, mobile phase pH and column oven temperature of glimepiride. [Click here to view] |
System suitability
To proceed, the validation of system suitability parameters was conducted in accordance with the guidelines provided in USP-NP chapters 621 and 1225. SST parameters for GLP were assessed across samples, diluent media, and mouse plasma. The RT of GLP was consistent in both samples at 2.81 and 2.82 minutes, each within a 5 minutes run time. The corresponding chromatograms are illustrated in Figure S2 a and b. Moreover, the system suitability parameters (SSTs), including NTP values of 27812 and 3019, tailing factors of 1.23 and 1.26, and % RSDs of 0.165 and 0.480, were observed and are shown in Table S1b.
Stock solution (diluent media and mouse plasma)
As suggested in the previous section, the stock samples were prepared individually. The initial stock concentration was 1 mg/ml, and the same sample was used for the determination of the LOD, LOQ, and linearity. Mouse blood samples were collected retro-orbitally from the BITS-PILANI animal facility, and additional plasma was separated for experimental use.
Sensitivity LOD and LOQ
The sensitivity of the method for detecting GLP was assessed by injecting lower concentrations to higher concentrations (0.0024–1.56 µg/ml) at seven different concentrations. The LOD and LOQ were found to be 0.066 µg/ml and 0.199 µg/ml, respectively. Similarly, in mouse plasma, the LOD and LOQ were found to be 0.193 µg/ml and 0.583 µg/ml, respectively. However, GLP could be quantified with accuracy and precision at the LOQ.
Matrix effect
The matrix effect was calculated using Equation 2; the maximum matrix effect was recorded at 81.9. Thus, we can conclude that the matrix effect is well within the acceptable range, as mentioned above. The calibration plots for both media are depicted in Figure 3.
Specificity
This experiment was validated according to the compendial method USP-1225, and the percent interference was calculated. GLP was identified at RT for 2.8 minutes with pure compound, and the same procedure was followed for the injected placebo and drug product (transdermal patch). The calculated interference was found to be 0.001%. The proposed analytical method was found to be suitable for the intended use (Fig. 2).
Linearity and range
The linearity and range were established as per ICH guidelines. In this experiment, the linearity study was performed individually in both diluent media, such as normal diluent media, and rat plasma. The r2 values were 0.9998 and 0.9998; the slopes were 62230.69 and 11258.7; and the intercepts were 8506.17 and −707.8, respectively. The quantification range was established from 25 to 100 µg/ml with accuracy and precision. The calculated result is depicted in Figure S5.
Method precision
The average recovery of GLP from diluent media was 100.00% ± 0.1% with a RSD of 0.001% for intraday and 101.79% ± 2.43% with an RSD of 0.023% for interday. In mouse plasma samples, the average recovery was 100% ± 0.707% with an RSD of 0.007%, and 100% ± 2.52% with an RSD of 0.025% (Table 2a). These results indicate that all method precision samples met the specifications of the USP and ICH standards. Therefore, it was concluded that the proposed analytical method is suitable for routine analysis and assay tests.
 | Table 2. Results summary of glimepiride (a) method precision (b) accuracy and recovery. [Click here to view] |
Accuracy
Through the accuracy study, the experimental results were validated at three different concentrations, namely, 150 µg/ml, 100 µg/ml, and 50 µg/ml, for both sample types: (a) diluent media and (b) mouse plasma. The average recoveries for n = 9 samples at these concentrations were 99.33% ± 3.09% RSD and 96.96% ± 5.46% RSD, respectively. In the case of the diluent media, the lower limit of individual recovery was 95.00%, followed by 90.00% for plasma. Similarly, the upper limits were 104.84% and 104.14%, respectively. Consequently, it can be inferred that the validated method is accurate and suitable for assessing the quality of GLP in drug products. The detailed findings are summarized in Table 2b.
Robustness
In this study, the analytical method was optimized using QbD. Although this parameter could have been excluded from the final validation, it was chosen for industrial application. Initially, a limited set of factors, including flow rate, NTP, and column oven temperature, were subjected to QbD. Subsequently, when robustness was validated, additional parameters, such as the mobile phase composition, sample cooler temperature, flow rate, and column oven temperature, were introduced. The mean recovery of GLP remained consistent at 100% ± 5% across all variable-robust parameters. However, variations in the RT were observed when the mobile phase composition ratio was altered. An increase of (+10%) led to an RT shift to 2.68 minutes, while a decrease of (–10%) resulted in an RT shift to 2.98 minutes. Nonetheless, the remaining SST parameters were either nearly equal or fell within ±10% of the acceptance range. The detailed experimental results are presented in Table 3a.
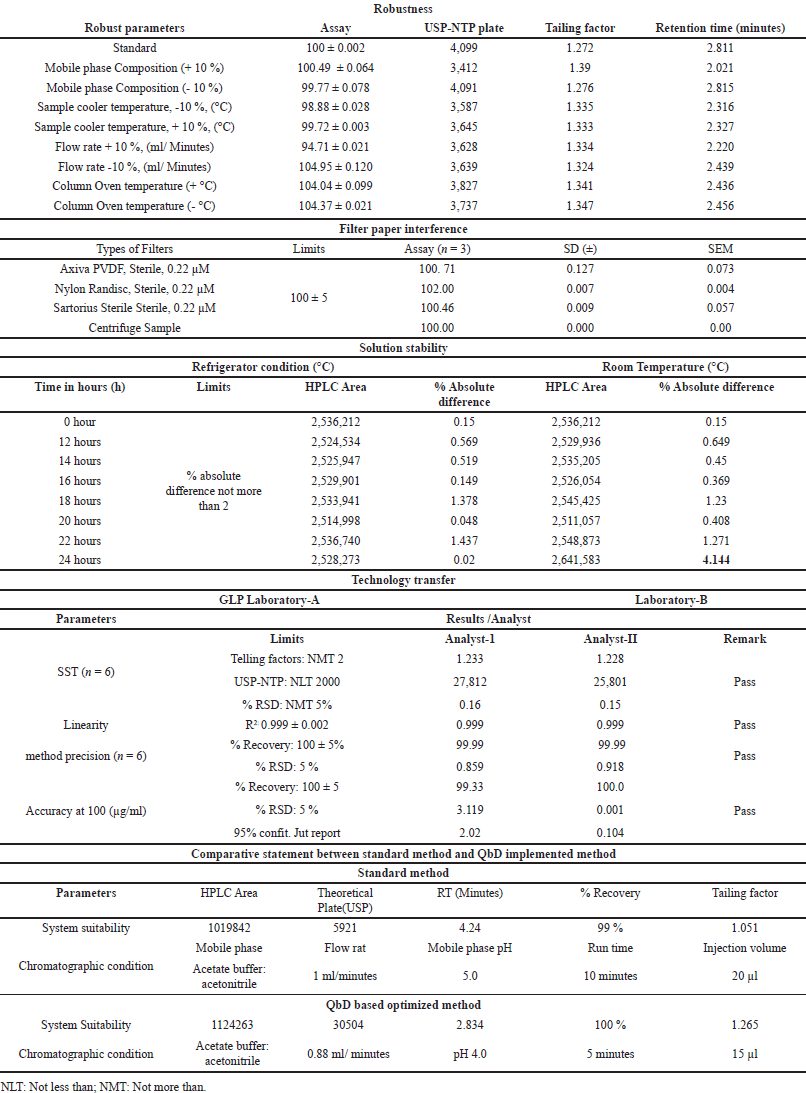 | Table 3. The results are summarized in the table: (a) Robustness (b) Filter paper interference (c) Solution stability (d) Technology transfer. [Click here to view] |
Filter paper interference
This parameter was validated using syringe filters from different manufacturers. The percentage recovery was determined individually for the syringe filter and centrifuge samples, yielding values of 100.71%, 102.00%, 100.46%, and 100.00%, respectively (Table 3b). The calculated results fell within the specified range of 95.0%–105.0%. Consequently, it was concluded that the selected syringe filters are suitable for assay method validation and routine analysis of samples in the quality control laboratory for GLP-loaded drug products. The detailed experimental findings are presented in Table 3b. Additionally, we were summarized the comparative statement between the standard method and QbD implemented method (Table 3e).
Solution stability
The solution stability was validated under two different temperature conditions, (a) room temperature and (b) refrigeration, at different time points. Throughout the experiment, percent recovery was calculated at each time point up to 36 hours, and the results were compared with those of a fresh standard sample at each time point. The percent absolute difference was calculated at each time point and is reported in Table 3c. The absolute difference was recorded for up to 36 hours, and the solution was found to be stable for up to 36 hours. Based on the experimental results, it was concluded that the quality control sample can be utilized for routine analysis for up to 24 hours at room temperature and up to 36 hours in a refrigerator (Table 3c).
Technology transfer
Both laboratory results were comparatively evaluated and calculated as percent RSD (n = 12 samples). The percent RSD was 0.888%, which met the acceptance criteria. The detailed results are summarized in Table 3d.
Industrial application
The percentages of GLP-loaded liposomes, GLP-loaded transdermal patches, GLP tablets (strength 2 mg), and GLP-loaded liposomes were 98.23%, 99.34%, and 93.34%, respectively. The corresponding chromatograms are shown in Figure S2 F–H. These findings were confirmed through three sets of experiments. Based on these experimental results, it can be concluded that the proposed analytical method has potential for industrial-scale use.
DISCUSSION
Method development is a critical part of the HPLC system. Our research findings revealed that the proposed analytical method is sensitive, accurate, and economical for industrial applications. HPLC detection plays a significant role in the sensitivity of the method; in this case, chromophore groups are present in the chemical structure of GLP (Fig. 1). The system suitability experimental results revealed that the present method qualifies as per the USP standard. In this experiment, GLP was eluted from the HPLC column at 2.8 minutes. Comparatively, the compound resolution and RT were shown to be better than those of existing methods [27,28]. In addition, Sebaiy et al. [29] reported that GLP can be separated from human blood plasma within 3 minutes. However, in our case, GLP was eluted from the column at RT for 2.8 minutes with mouse plasma (Fig. S2a and b). The developed method has shown equal potential for bioanalytical applications. The extraction of active compounds from mouse plasma is a significant challenge due to the matrix effect and poor accuracy. Several strategies can be employed to mitigate the impact of matrix effects in HPLC analysis. Sample preparation techniques like solid-phase extraction or liquid–liquid extraction can be used to clean up plasma samples prior to HPLC analysis. Additionally, using calibration curves derived from matrix-matched standards can also improve accuracy by accounting for the specific matrix effects present in the biological samples [30]. However, in the present study, we used the liquid?liquid extraction method to counter the above obstacles [31].
During analytical method development, the selection of solvent, mobile phase buffer pH, and HPLC column play a major role in the separation of the analyte [32]. In this case, we used universal solvents such as Milli-Q- water, acetonitrile, and methanol for the intended use. The GLP peak was separated from the HPLC column after 5 minutes. Furthermore, the developed method was optimized using a Box?Behnken design with three levels of factors, which increased the quality of the method and minimized the chemical cost (Fig. 2). This technology was implemented by the US FDA in 2004 and has been globally accepted by the International Thoracic Committee (ICH) since 2005 [33]. Recently, Athar Shamim et al. [34] reported an analytical method for the simultaneous estimation of rutin and ciprofloxacin by the QbD approach. His findings suggested that the developed RP-HPLC method can be successfully applied to analyze rutin and ciprofloxacin-loaded liposomal nano formulations prepared by the thin-film hydration technique. The QbD-based HPLC method is foundational in advancing pharmaceutical applications, particularly in developing formulations that require enhanced solubility and bioavailability. This approach not only streamlines the analytical process but also aligns with the industry’s growing emphasis on sustainable and patient-centric drug delivery technologies. Thus, our proposed optimized HPLC method could be used for estimation of GLP from various pharmaceutical dosages regimes such as solid oral formulation, liposomal formulation and transdermal patch (Fig. S2 D–F).
On the other hand, numerous analytical methods have reported simultaneous estimation methods. However, when considering an industrial scale, most methods still need to meet industrial specifications. However, in this study, we used a reference standard to meet the industrial requirements. Numerous research papers validating various aspects of the method, including system suitability, LOD and LOQ, linearity, range, specificity, precision, accuracy, and robustness, have been published. However, it is important to note that academic research has yet to encompass studies related to solution stability and filter paper interference [35,36]. Solution stability is not an integral part of analytical method validation as per ICH Q2 (R1). However, pharmaceutical companies must work with routine samples from 24-hour working shifts; therefore, it is mandatory to validate this parameter for 24 hours or more. Similarly, filter paper interference is a new approach for analytical method validation, but it has yet to be considered by regulatory agencies. The method was tested in various pharmaceutical dosages form such as liposome, solid oral formulation, transdermal patch and found satisfactory. However, this same method could be used for the quantification of glimepiride form various pharmaceutics dosages form [37–40].
CONCLUSION
A successful analytical method was developed and validated. Our findings revealed that the proposed analytical method is sensitive and economical at the industrial scale and capable of estimating bioanalytical samples. The key novelty of this method is its short run time and minimal solvent waste, leading to cost savings. The developed method is reproducible at both laboratories, the originator and the receiver. It can be concluded that the method is reproducible at the industrial scale. The application of this method can be explored further by extending it to a pharmacokinetic study of GLP-loaded transdermal patches. Thus, the developed method could be used for both analytical and bioanalytical purposes at the industrial scale. The proposed analytical method is not only cost-effective but also precise and less time-consuming. Consequently, we can affirm that the proposed method is suitable for quantifying solid, liquid, and topical formulations for industrial use.
AUTHOR DECLARATION
The authors have no financial or proprietary interests in any material discussed in this article.
ACKNOWLEDGMENTS
The authors acknowledge CSIR-CDRI, Lucknow, for providing infrastructure support for this research. Furthermore, the authors would like to acknowledge the Central Analytical Facility (CAL Lab-1) and Centre for Human Diseases and Research (CHDR), Birla Institute of Technology and Sciences-Pilani Hyderabad campus, for providing the laboratory facility to prove this hypothesis.
AUTHOR CONTRIBUTIONS
Mr. Abhiram Kumar: Ideation, experimentation and final manuscript drafting; Ms. Chhavi Dhiman: Performed method precision experiments at the receiver site; Mr. Madhaw Kumar: Wet lab analysis and data recording; Prof. N. Kannappan: Review and editing. Mr. Deepak Sharma: Review and editing; Dr. Manish Kumar Chourasia Provided the laboratory facilities for analytical method development and validation at the originator site. Prof. Kumar Pranav Narayan provided us with a laboratory facility and guided us to carry out this research project at the receiver site. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
The Institutional Animal Ethics Committee of the Department of Biological Sciences, Birla Institute of Technology and Sciences, Pilani, Hyderabad Campus, India, approved the study protocol with approval number: BITS-HYD/IAEC/2020-31.
DATA AVAILABILITY
The datasets generated and/or analyzed during the current study are not publicly available due to privacy and security but are available from the corresponding author upon reasonable request
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. EMA. Quality by design. [cited 2024 Feb 25]. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/research-and development/qualitydesign#:~:text=One%20of%20the%20goals%20of,is%20’right%20first%20time’
2. Kumar A, Kumar M, Singh R, Upadhyay P, Mukherjee A. Quality by design (QbD) aided formulation optimization of amlodipine besylate oral thin film. Indian J Pharm Educ Res. 2024;58(2s):s444–52. CrossRef
3. ICH. Pharmaceutical development Q8(R2). Geneva, Switzerland: International Council For Harmonization; 2009 [cited 2024 Oct 30]. Available from: 2024.(https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
4. WHO. Diabetes in India. Geneva, Switzerland: WHO; 2024 [cited 2023 Aug 22]. Available from: https://www.who.int/india/health-topics/mobile-technology-for-preventing-ncds
5. Chan JCN, Lim LL, Wareham NJ, Shaw JEee. The Lancet commission on diabetes: using data to transform diabetes care and patient lives. Lancet 2021;396(10267):2019–82. CrossRef
6. Collaborators GBDD. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023;402(10397):203–34. CrossRef
7. ICH. Pharmaceutical development Q8(R2). In: harmonization Icf, editor. Geneva, Switzerland: International council for harmonization; 2009.
8. US-FDA. Guidance for industry, Q8, Q9, & Q10. In: US-FDA, editor. United States of America: U.S. Department of Health and Human Services; July 2012: pp. 1–17.
9. Patel KY, Dedania ZR, Dedania RR, Patel U. QbD approach to HPLC method development and validation of ceftriaxone sodium. Futur J Pharm Sci. 2021;7(1):141. CrossRef
10. Sangshetti JN, Deshpande M, Zaheer Z, Shinde DB, Arote R. Quality by design approach: regulatory need. Arab J Chem. 2017;10:S3412–25. CrossRef
11. ICH. Analytical procedure development Q14. Geneva, Switzerland: International Council of Harmonization; 2022: pp. 1–61.
12. US-FDA. Analytical procedures and methods validation for drugs and biologics. Silver Spring, MD: U.S. Department of Health and Human Services Food and Drug Administration; 2015 [cited 2024 Oct 30]. 1–13 pp. Available from: https://www.fda.gov/files/drugs/published/Analytical-Procedures-and-Methods-Validation-for-Drugs-and-Biologics.pdf
13. Marie AA, Hammad SF, Salim MM, Elkhodary MM, Kamal AH. Deduction of the operable design space of RP-HPLC technique for the simultaneous estimation of metformin, pioglitazone, and glimepiride. Sci Rep 2023;13(1):4334. CrossRef
14. USP-NF. Chromatography. The United States Pharmacopeia – National Formulary. Vol 47. United State of America: The United States Pharmacopeial Convention; 2022.
15. Maurya P, Kumar A, Singh S, Nisha R, Pal RR, Chourasia MK, et al. Bio-analytical method development for estimation of levofloxacin: application in estimation of drug in nano-formulations and pharmacokinetic studies. Indian J Pharm Educ Res. 2021;55(3s):S814–24. CrossRef
16. Qazi F, Shoaib MH, Yousuf RI, Fahad S, Muhammad IN, Kamran , et al. QbD based eudragit coated meclizine HCl immediate and extended release multiparticulates: formulation, characterization and pharmacokinetic evaluation using HPLC-Fluorescence detection method. Sci Rep 2020;10(1):14765. CrossRef
17. ICH. Validation of analytical procedures. Geneva, Switzerland: ICH; 2023 [cited 2024 Oct 30]. Available from: https://www.ich.org/page/quality-guidelines.
18. Sha’at M, Spac AF, Stoleriu I, Bujor A, Cretan MS, Hartan M, et al. Implementation of QbD approach to the analytical method development and validation for the estimation of metformin hydrochloride in tablet dosage forms by HPLC. pharmaceutics 2022;14(6). CrossRef
19. Taverniers I, De Loose M, Van Bockstaele E. Trends in quality in the analytical laboratory. II. Analytical method validation and quality assurance. TrAC, Trends Anal Chem. 2004;23(8):535–52. CrossRef
20. Regulations F-CoF. Title 21, Volume 4, CITE: 21CFR211.194. CFR-Code of Federal Regulations Title 21; 2023.
21. Martinez-Ortega A, Herrera A, Salmeron-Garcia A, Cabeza J, Cuadros-Rodriguez L, Navas N. Study and ICH validation of a reverse-phase liquid chromatographic method for the quantification of the intact monoclonal antibody cetuximab. J Pharm Anal 2016;6(2):117–24. CrossRef
22. Okamoto M. Assay validation and technology transfer: problems and solutions. J Pharm Biomed Anal 2014;87:308–12. CrossRef
23. USP-NF. Transfer of analytical procedures. The United States Pharmacopeia – National Formulary. Rockville, MD: The United States Pharmacopeial Convention; 2023. Vol. 40. CrossRef
24. Kumar A, Rana R, Saklani R, Madhaw K, Pavan KY, Amrendra T, et al. Technology transfer of a validated rp-hplc method for the simultaneous estimation of andrographolide and paclitaxel in application to pharmaceutical nanoformulation. J Chromatogr Sci. 2023. CrossRef
25. USP-NF. Analytical Interpretation and treatment. The United States Pharmacopeia – National Formulary. Rockville, MD: United State of America: The United States Pharmacopeial Convention; 2019. Vol 35. pp 436–48.
26. Srivastava S, Kumar A, Yadav PK, Kumar M, Mathew J, Pandey AC, et al. Formulation and performance evaluation of polymeric mixed micelles encapsulated with baicalein for breast cancer treatment. Drug Dev Ind Pharm 2021;47(9):1512–22. CrossRef
27. Samala S, Tatipamula SR, Veeresham C. Determination of Glimepiride in rat serum by Rp-Hplc method. Am J Anal Chem. 2011;02(02):152–7. CrossRef
28. Dash RN, Mohammed H, Humaira T. An integrated Taguchi and response surface methodological approach for the optimization of an HPLC method to determine glimepiride in a supersaturatable self-nanoemulsifying formulation. Saudi Pharm J. 2016;24(1):92–103. CrossRef
29. Sebaiy MM, El-Adl SM, Baraka MM, Hassan AA. Rapid RP-HPLC method for simultaneous estimation of metformin, pioglitazone, and glimepiride in human plasma. Acta Chromatogr. 2019;32(1):16–21. CrossRef
30. Kim T, Han DG, Yoon IS. A simple and sensitive high-performance liquid chromatographic method combined with fluorescence detection for bioanalysis of scopoletin in rat plasma: application to a pharmacokinetic study. Biomed Chromatogr. 2024;38(10):e5959. CrossRef
31. Parthasarathy S, Ramanathan S, Ismail S, Adenan MI, Mansor SM, Murugaiyah V. Determination of mitragynine in plasma with solid-phase extraction and rapid HPLC-UV analysis, and its application to a pharmacokinetic study in rat. Anal Bioanal Chem 2010;397(5):2023–30. CrossRef
32. Kataoka H. Chapter 1—sample preparation for liquid chromatography. In: Fanali S, Chankvetadze B, Haddad PR, Poole CF, Riekkola M-L, editors. Liquid Chromatography (Third Edition). Amsterdam, The Netherlands: Elsevier; 2023. Vol 2. pp 1–48. CrossRef
33. Tome T, Žigart N, ?asar Z, Obreza A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: overview and recent advances. Org. Process Res. Dev. 2019;23(9):1784–802. CrossRef
34. Shamim A, Ansari MJ, Aodah A, Iqbal M, Aqil M, Mirza MA et al. QbD-engineered development and validation of a RP-HPLC method for simultaneous estimation of rutin and ciprofloxacin HCl in bilosomal nanoformulation. ACS Omega 2023;8(24):21618–27. CrossRef
35. Guerrero-Hurtado E, Gutierrez-Docio A, Fiedorowicz R, Molla E, Reglero G, Prodanov M. Why proanthocyanidins elute at increasing order of molecular masses when analysed by normal phase high performance liquid chromatography? Considerations of use. J Chromatogr A. 2023;1696:463957. CrossRef
36. Vasileiadou A, Karapanagiotis I, Zotou A. Development and validation of a liquid chromatographic method with diode array detection for the determination of anthraquinones, flavonoids and other natural dyes in aged silk. J Chromatogr A. 2021;1651:462312. CrossRef
37. Rana R, Mishra K, Tripathi S, Gupta AK, Tiwari AK, Yadav PK, et al. Simultaneous estimation of rutin and donepezil through RP-HPLC: implication in pharmaceutical and biological samples. Bioanalysis. 2024;16(11):557–67. CrossRef
38. Maurya P, Saklani R, Singh S, Nisha R, Mishra N, Singh P, et al. Effective uptake of folate-functionalized ethionamide-loaded hybrid system: targeting alveolar macrophages. Nanomedicine. 2022;17(24):1819–31. CrossRef
39. Kumar A, Sakhare K, Bhattacharya D, Chattopadhyay R, Parikh P, Narayan KP, et al. Communication in non-communicable diseases (NCDs) and role of immunomodulatory nutraceuticals in their management. Front Nutr. 2022 Sep 21;9:966152.
40. Sharma M, Joshi J, Chouhan NK, Talati MN, Vaidya S, Kumar A. Liposome-A comprehensive approach for researchers. Mol Pharmacol. 2020.
SUPPLEMENTARY MATERIAL
The supplementary material can be accessed at the journal’s website: Link here https://japsonline.com/admin/php/uploadss/4482_pdf.pdf.