
INTRODUCTION
Orlistat (C29H53NO5, (S)-((S)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl) tridecan-2-yl) 2-formamido-4-methylpentanoate, Molecular wt. 495.745 g/mol) (Fig. 1), a lipase inhibitor, reduces fat absorption by inhibiting pancreatic lipase activity. Clinical trials affirm its efficacy in promoting weight loss and improving metabolic parameters. Orlistat has been shown to exert beneficial effects on cardiovascular risk factors, such as dyslipidemia and hypertension, making it a valuable adjunctive therapy in the management of obesity-related comorbidities [1].
Orlistat, a lipase inhibitor, acts by selectively targeting the pancreatic lipase enzymes within the gastrointestinal tract, thereby impairing the hydrolysis of dietary triglycerides into absorbable free fatty acids and monoacylglycerols. Consequently, Orlistat reduces the absorption of dietary fats by approximately 30%, leading to a decrease in caloric intake and subsequent weight loss [2–3]. The therapeutic efficacy and safety profile of Orlistat has been extensively evaluated in numerous clinical trials and observational studies. These investigations have demonstrated its effectiveness in promoting weight loss, improving metabolic parameters, and reducing the risk of obesity-related complications. Furthermore, Orlistat has been shown to exert beneficial effects on cardiovascular risk factors, such as dyslipidemia and hypertension, making it a valuable adjunctive therapy in the management of obesity-related comorbidities [4].
The extensive literature on Orlistat highlights various analytical methods for its estimation, encompassing HPTLC [5], thin layer chromatography [6], RP-HPLC [7,8], HPLC/MS [9], and LC-MS/MS [10–21]. However, despite the availability of numerous LC-MS/MS methods, none have employed the design of experiments (DOEs) approach for both method development and optimization in quantifying Orlistat in human plasma. In this study, we introduce a novel LC-MS/MS method for the precise quantification of Orlistat in human plasma, leveraging the DOE approach. By systematically varying multiple factors simultaneously, the DOE method allows for the efficient exploration of the interaction effects between parameters, resulting in an optimized analytical method with enhanced robustness and accuracy. This innovative approach fills a significant gap in the existing literature and offers a robust and efficient method for Orlistat quantification, ensuring its reliable application in clinical and pharmaceutical research.
 | Figure 1. Chemical structure of Orlistat. [Click here to view] |
MATERIALS AND METHODS
Reagent and chemicals
Jigs Chemical Limited, Ahmedabad, India, provided the Orlistat and Orlistat-D5. Formic acid, methanol, and acetonitrile are of analytical grade and purchased from SD Fine Chem Ltd, Hyderabad, India. The built-in water supply of the Milli-Q® RO system was utilized for preparing the mobile phase as well as washing solvents.
LC-MS/MS instrumentation
This study used an Agilent 1260 series HPLC system (Agilent Technologies, Santa Clara, CA, USA) and an API 3,200 mass spectrometer (AB SCIEX, Framingham, MA, USA) with a turbo electrospray interface. Orlistat and Orlistat-D5 were quantified using the mass spectrometer in MRM mode with positive electrospray ionization (ESI+). Orlistat and Orlistat-D5 were separated using a reversed-phase Phenomenex-C18 column (2.1 mm × 50 mm, 5 µ) and an isocratic solvent solution (methanol, acetonitrile, 0.1% formic acid, 65:20:15 (V/V/V) at 0.8 ml/minute). The autosampler was kept at 10°C during analysis. Instrument control and data analysis were done with Analyst 1.5.2 (Applied Biosystems, Foster City, CA, USA).
Mass instrument parameters
Both Orlistat and Orlistat-D5 underwent positive ionization mode utilizing the multiple reaction monitoring (MRM) mode on the mass spectrometer. The flow rates of the drying gas and sheath gas were kept at 15.0 and 4.0 l/minute, respectively, while a capillary voltage of 2.5 kV was applied. Every transition was given a dwell duration of 200 milliseconds. The impact energy and fragment voltage for both Orlistat and Orlistat-D5 were configured to 25 eV and 90 V, respectively. The pressure of the nebulizer was consistently maintained at 25.0 psi. Both analytes were subjected to an optimized collision energy voltage of 25 V, while the source temperature was set at 500°C. The most prevalent product ions detected were at m/z 142.08, originating from the precursor’s ion at m/z 496.4 for Orlistat, and at m/z 147.07, originating from the precursor’s ion at m/z 501.3 for Orlistat-D5.
Linearity standards
Dissolving 100 mg of Orlistat in 100 ml mobile phase produced a 1mg/ml stock solution. Orlistat standard solution was spiked into blank plasma to establish calibration standards at 1.2, 5.6, 27.0, 75.0, 145.0, 221.0, 305.0, and 392.0 ng/ml.
Quality standards
The calibration standard solutions set the lowest (LQC), medium (MQC), and highest (HQC) quality control standards. These quality control (QC) samples were calibrated to 6.0, 196.0, and 290.0 ng/ml for LQC, MQC, and HQC. The prepared solutions were kept at −20°C until the completion of analysis.
Sample preparation
To prepare the sample solution, 250 µl plasma and 100 µl Internal Standard (IS) (1 µg/ml) were mixed and vortexed for 2 minutes. Orlistat and IS were extracted in 5.0 ml ethyl acetate. The solution was centrifuged for 30 minutes at 4,500 rpm/minute. Lyophilizers dried the organic phase after centrifugation. The final product was solubilized in 250 µl of mobile phase and placed in pre-labeled vials. In the auto-sampler, the vials were infused into the LC-MS/MS.
Design of experiments
The current investigation employs a design of experiments (DOEs) framework to formulate a simple and robust LC-MS/MS methodology for the precise quantification of Orlistat within biological matrices. The selection of the Box-Behnken experimental design for method development is predicated upon its notable merits encompassing optimal resource utilization, equilibrium of factor levels, heightened resilience to extraneous influences, and proficiency in the discernment of quadratic effects. This design facilitates the comprehensive exploration of factor permutations, concomitantly mitigating procedural variance, thus accommodating the constraints inherent in a restricted experimental domain. The application of response surface modeling inherent to this design furnishes predictive insights into response patterns, thereby affording the identification of paramount parameter configurations that optimize the methodology.
Within this paradigm, four pivotal experimental variables emerged as determinative, thereby being established as autonomous factors: specifically, capillary voltage (A), cone voltage (B), desolvation temperature (C), and collision energy (D). The establishment of the operational scope of these variables was undertaken by anchoring them within empirically derived minimum and maximum levels. Specifically, Capillary Voltage was delimited within the range of 1.5 kV to 4.5 kV, Cone Voltage spanned from 10 V to 35 V. Desolvation temperature encompassed the interval of 350°C–550°C, and the Collision energy was confined within 10 eV–20 eV.
Given the paramount significance of analyte quantification within the ambit of bioanalytical methodology employing Mass spectrometric detection, the present investigation adopts the response area of orlistat (R1) and the IS (R2) as pivotal response variables meriting optimization efforts. A total of 29 different experiments were conducted according to the Box–Behnken design. The data table containing the factors at different levels and their measured responses after the experimental runs were enumerated in Table 1.
Method validation
The developed methodology was validated by assessing its selectivity, stability, specificity, linearity, matrix effect, precision, recovery, and accuracy (FDA, 2001; EMEA, 2011).
RESULTS
Optimization of LC-MS/MS method
We employed the MRM in positive mode with Orlistat to improve the mass determination sensitivity and specificity. Since ESI was likely the main source of ionization for LC-MS/MS, standard solutions were injected into the mass instrument with the help of a syringe pump to determine precursor and product ions. The detection ions, Orlistat, and IS’s product ions have mass spectra at m/z 142.08 and 147.07. To increase mass response, mass spectrum surrounding factors such as ion-spray voltage, temperature, capillary voltage, heater gas, collision gas, curtain gas, and nebulizer gas were set.
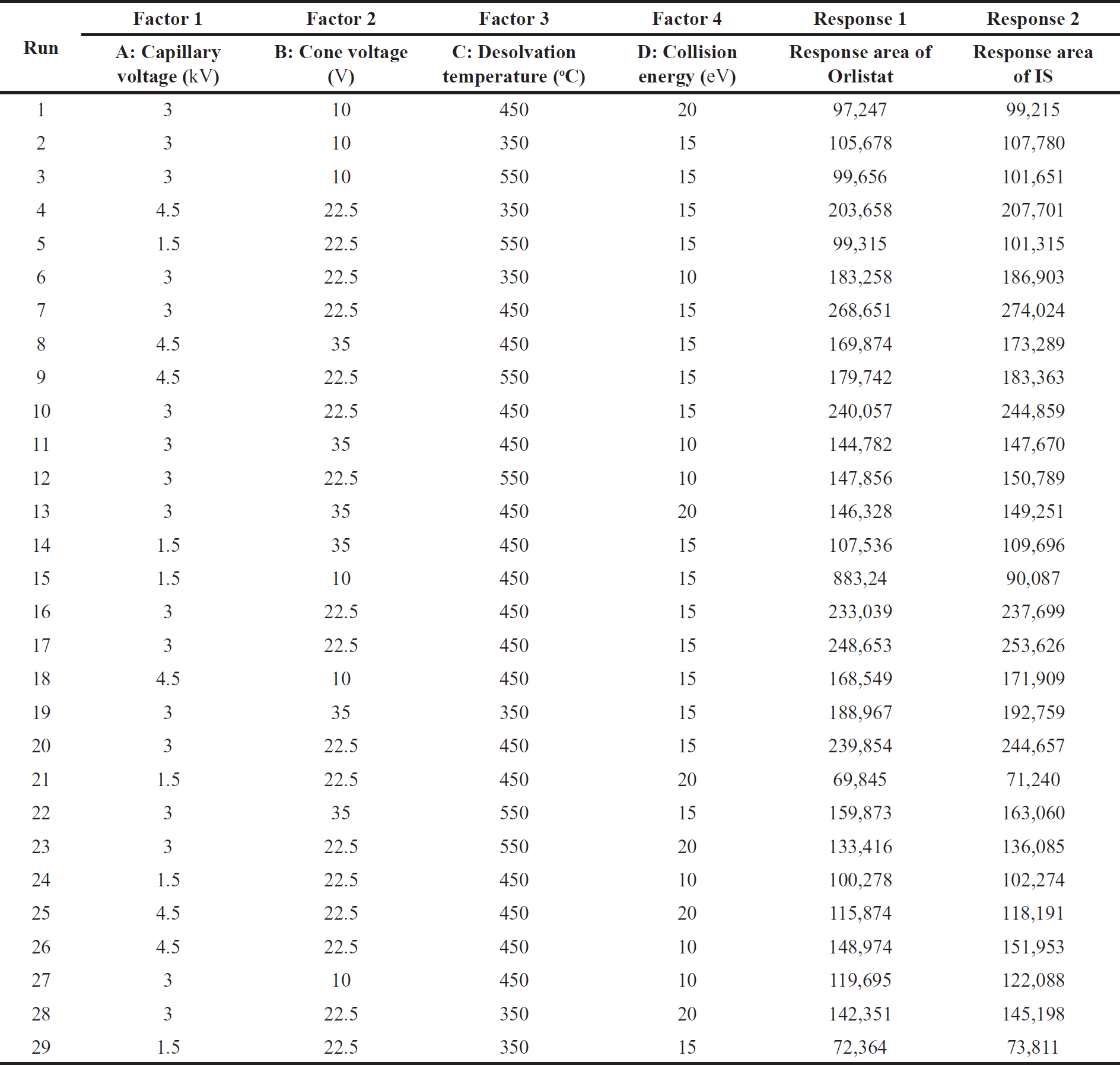 | Table 1. Summary of factors at different levels and their measured response. [Click here to view] |
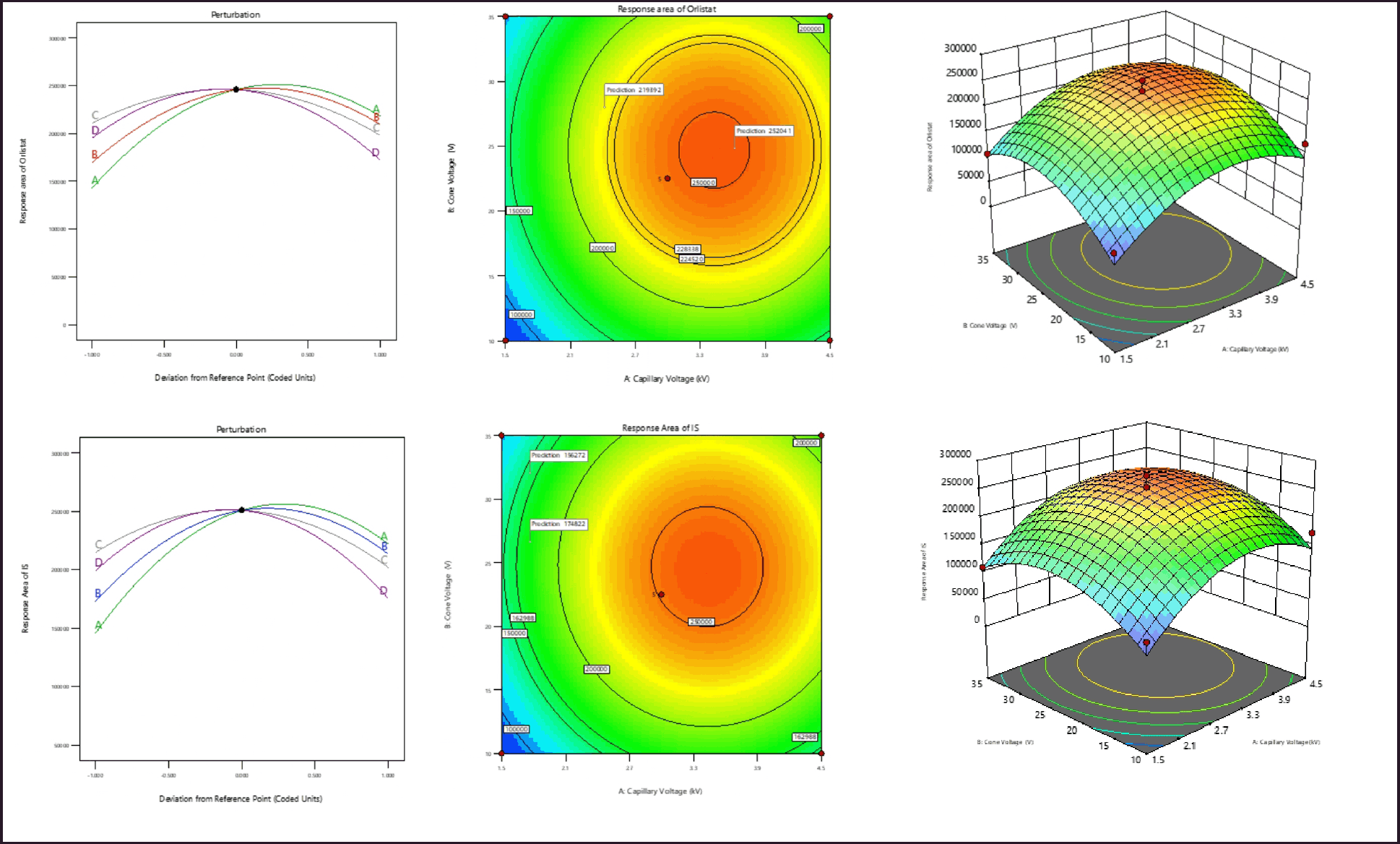 | Figure 2. (a) Perturbation plot, contour plot, and 3D surface plots of response area of Orlistat. (b) Perturbation plot, contour plot, and 3D surface plots of response area of internal standard. [Click here to view] |
Selection of IS
In this investigation, Orlistat-D5 was selected as the IS because, as shown in the method validation data, no clear interferences were seen at the analyte and IS retention durations. Its chromatographic behavior, ionization, extraction efficacy, and retention action were comparable to Orlistat’s.
Design of experiments
Response of Orlistat (R1)
The quadratic model created for the response variable -R1 is deemed significant, as evidenced by the F-value of 26.09. In the present model, it was seen that the variables A, B, A2, B2, C2, and D2 exhibited statistical significance, as indicated by their respective p-values being less than 0.005. The mathematical model suggests that the variables Capillary Voltage (A) and Cone Voltage (B) have a significant impact on the reaction of Orlistat (R1). There is a strong correlation between the observed values of R1 and the projected values. The model’s signal-to-noise ratio (S/N) of 22.62 suggests that the signal is sufficient. The impact of an individual variable on R1 is visualized through the utilization of a perturbation plot, contour plot, and 3D surface plot, as illustrated in Figure 2a. The examination of the response plots and regression equation reveals a clear indication that variables A and B exert a positive influence on R1. At elevated levels, all four factors have demonstrated a statistically significant quadratic impact on R1.
Response area of IS (R2)
The significance of the quadratic model developed for the response variable -R2 is indicated by the F-value of 26.09. In the current model, the variables A, B, A2, B2, C2, and D2 were statistically significant, as indicated by p-values less than 0.005. The mathematical model shows that the variables Capillary Voltage (A) and Cone Voltage (B) have a significant effect on the Ion Source’s (R2) reaction. There exists a substantial correlation between the observed and predicted values of R2. The signal-to-noise ratio (S/N) of 22.63 indicates that the signal strength is adequate. Figure 2b illustrates the effect of a single variable on the coefficient of determination (R2) through the use of the Perturbation plot, Contour plot, and 3D Surface plot. Upon inspection of the response diagrams and regression equation, it is evident that variables A and B have a positive effect on the coefficient of determination (R2). All four variables have statistically significant quadratic effects on the coefficient of determination (R2) at elevated levels. Figure 3 depicts the overlay diagram illustrating the analysis of the study’s responses.
Method validation
Specificity
Blank plasma samples from six human plasma batches were spiked with Orlistat at lower limit of quantification (LLOQ) and IS to test specificity. Figure 4 shows IS and Orlistat retention times of 2.93 minutes. Orlistat analysis showed no matrix material or IS interference, and all interfering peaks were less than 20% of LLOQ samples [10,11].
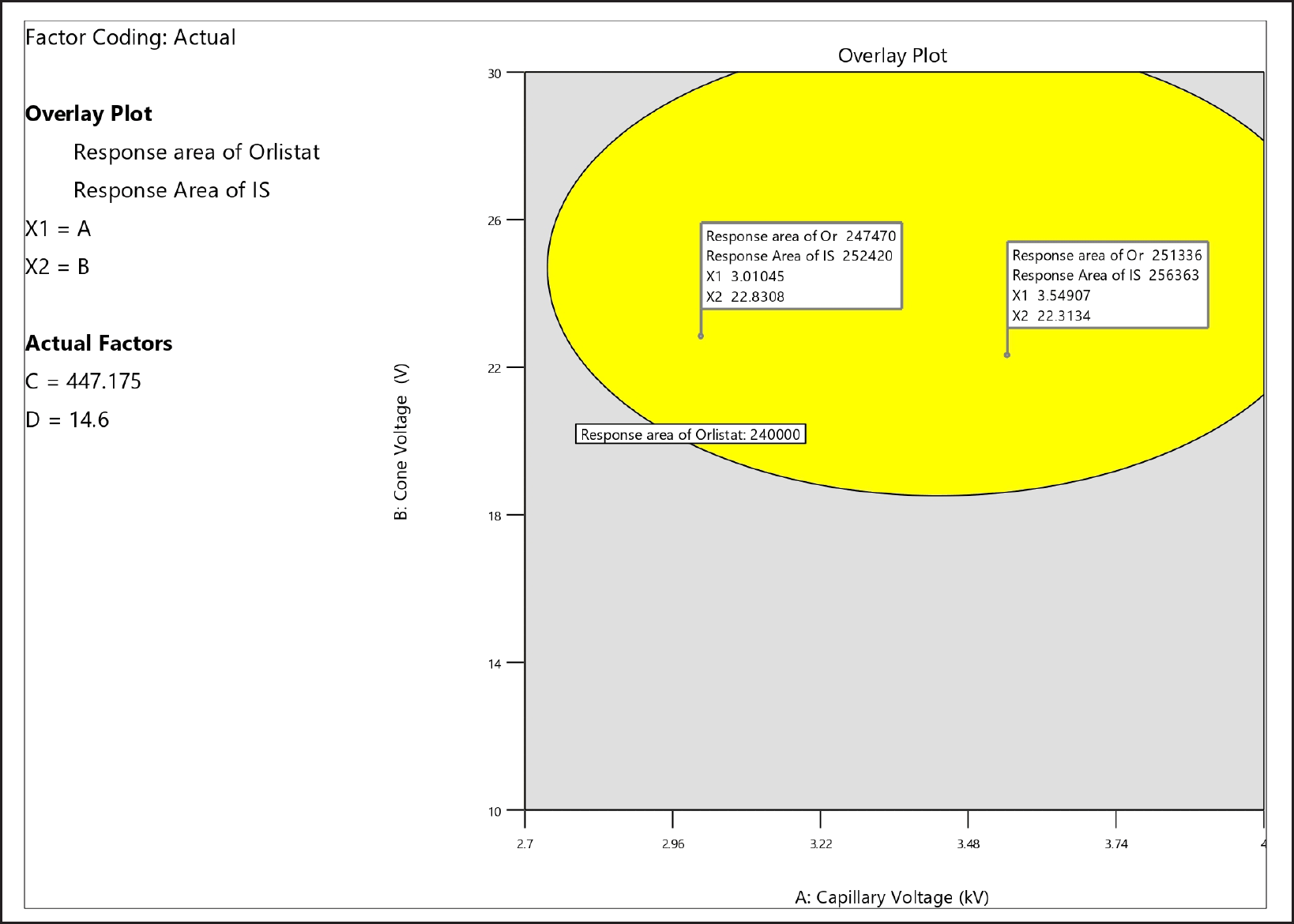 | Figure 3. Overlay plot of the responses. [Click here to view |
 | Figure 4. (A) Blank plasma (B) LLOQ sample chromatograms. [Click here to view] |
Linearity and sensitivity
The Orlistat linearity method was developed and showed good linearity from 1.2 to 392.0 ng/ml (Table 2). Linearity graphs were created using Orlistat peak area ratios to IS vs concentrations (x) using the 1/C2 weighting factor. Figure 5 shows the calibration graphs’ technique linearity equation, y = 0.003546 x – 0.004133, with a r2 value of 0.9999. Orlistat could be accurately quantified in plasma samples using its LLOQ of 1.2 ng/ml (signal-to-noise ratio > 10) [12,13,14].
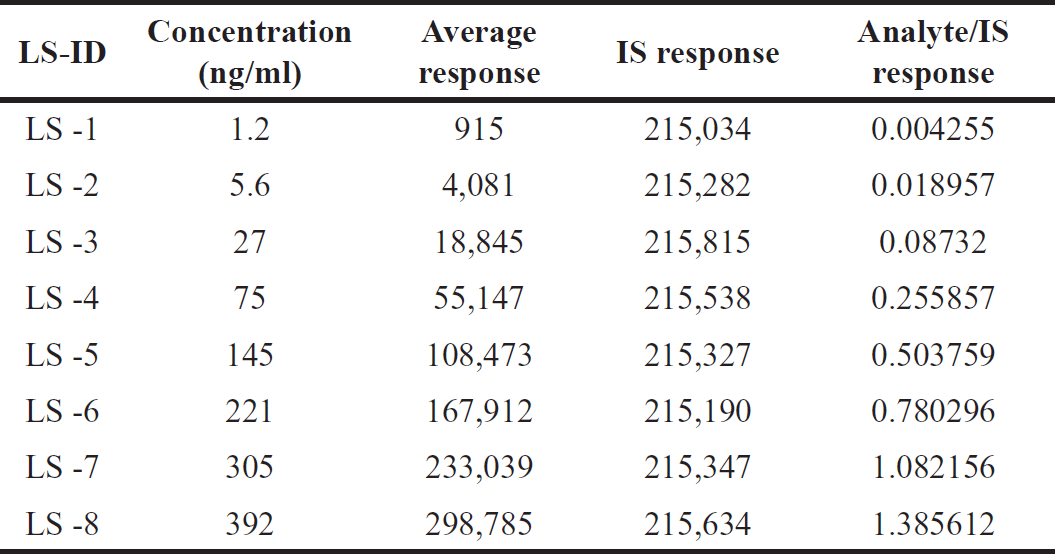 | Table 2. Linearity standard solutions for Orlistat. [Click here to view] |
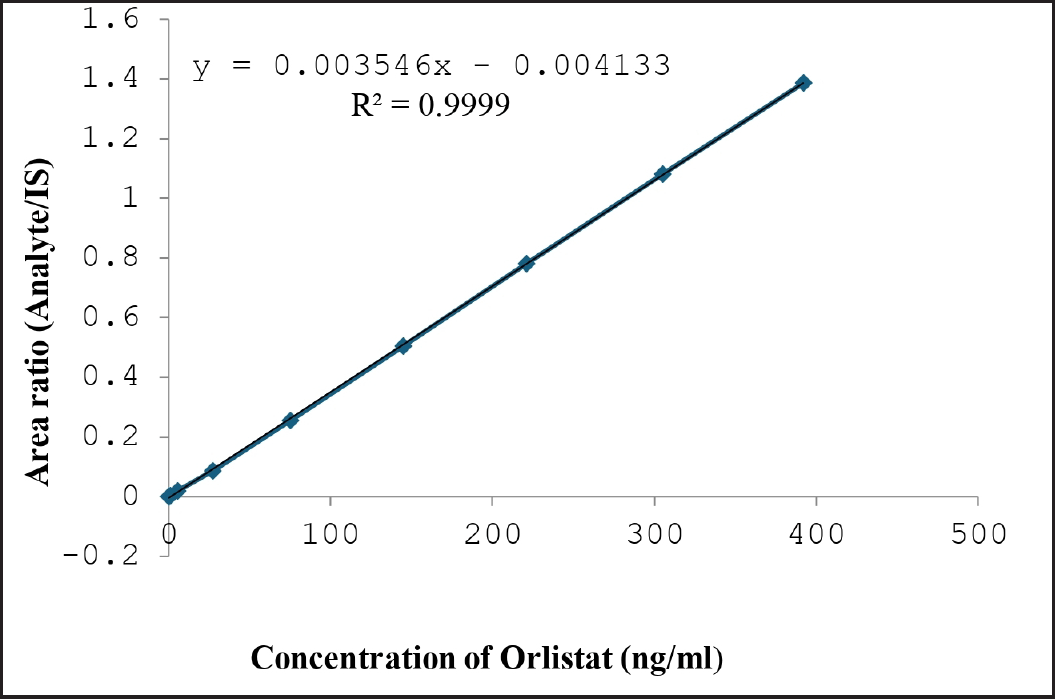 | Figure 5. Linearity of Orlistat. [Click here to view] |
Accuracy and precision
Six plasma samples spiked with Orlistat at HOQ, MQC, LQC, and LLQC levels were examined in one batch and three consecutive batches to determine intra- and inter-batch precision and accuracy. Table 3 enumerates the precision and accuracy of Orlistat quantification. The accuracy % RSD values for inter- and intra-batch analyses ranged from 2.84% to 5.32% [15,16].
Extraction recovery
Effective pretreatment was performed on biological material before evaluation. The peak area ratio of HQC, medium quality control (MQC), and low-quality control (LQC) level Orlistat solutions (n = 6) to extracted spiking samples at corresponding concentration levels were used to measure extraction recovery. Comparing the peak area ratio of quality control plasma sample solutions (n = 6) to spiked human plasma sample solutions at different concentrations indicated the IS extraction recovery. The mean extraction recovery of Orlistat was 103.62% at low QC, 96.14% at medium, and 95.73% at high. At 175 ng/ml, IS extraction recoveries averaged 99.05%. The findings are displayed in Figures 6–8 and Table 4 [15].
Matrix effect
Since co-eluting matrix constituents have the potential to either decrease or boost the ionization process in the mass system, it is possible that the blank matrix will not exhibit any discernible responses. As a consequence, the IS normalized matrix factor was calculated in eight different sources of human plasma. These sources included two batches that were hemolytic and two batches that were lipemic. Table 5 presented the findings, which showed that the average IS normalized matrix factor for all the analytes that were present fell within the range of 0.94–1.09, with a % RSD that was lower than 4.38 on average [12].
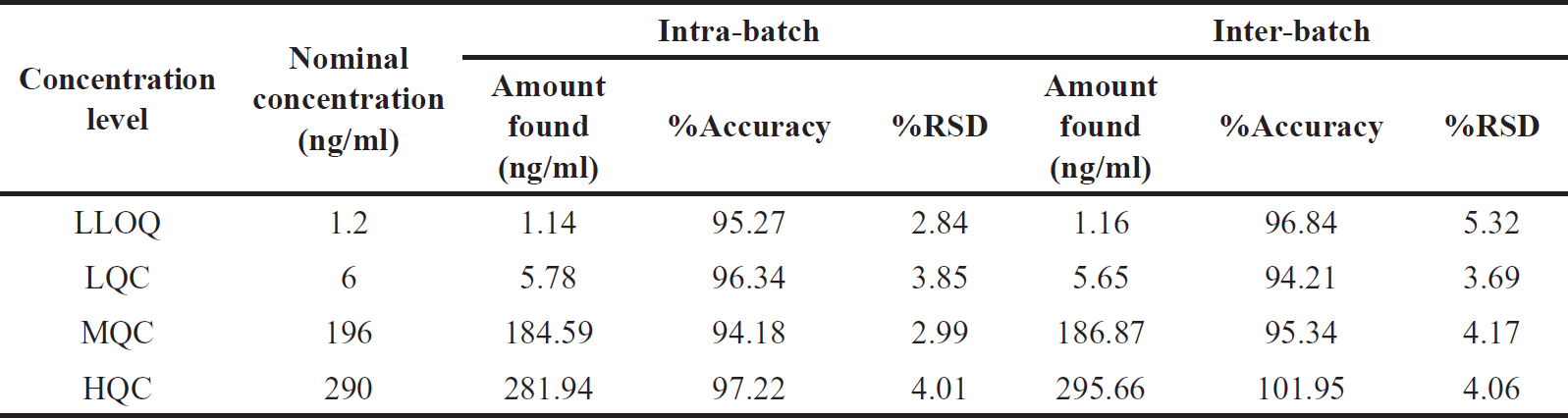 | Table 3. Orlistat precision and accuracy for Inter-batch and intra-batch. [Click here to view] |
 | Figure 6. Orlistat chromatogram at LQC standard. [Click here to view] |
 | Figure 7. Orlistat chromatogram at MQC standard. [Click here to view] |
 | Figure 8. Orlistat chromatogram at HQC standard. [Click here to view] |
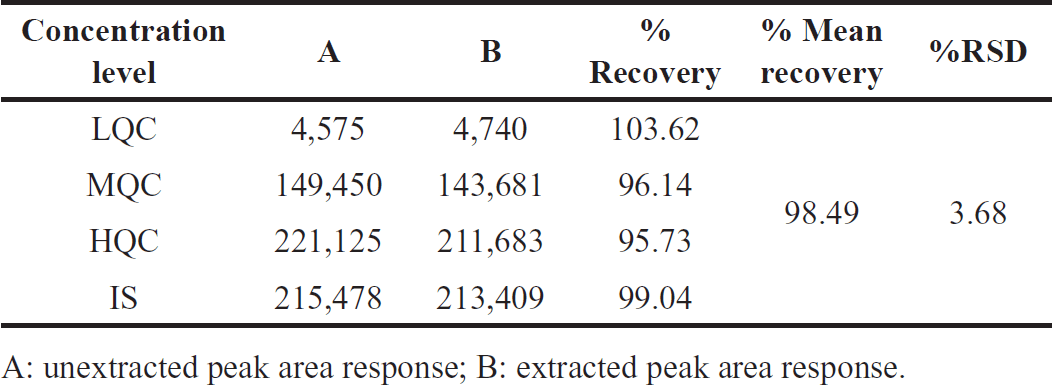 | Table 4. Orlistat and IS extraction recoveries. [Click here to view] |
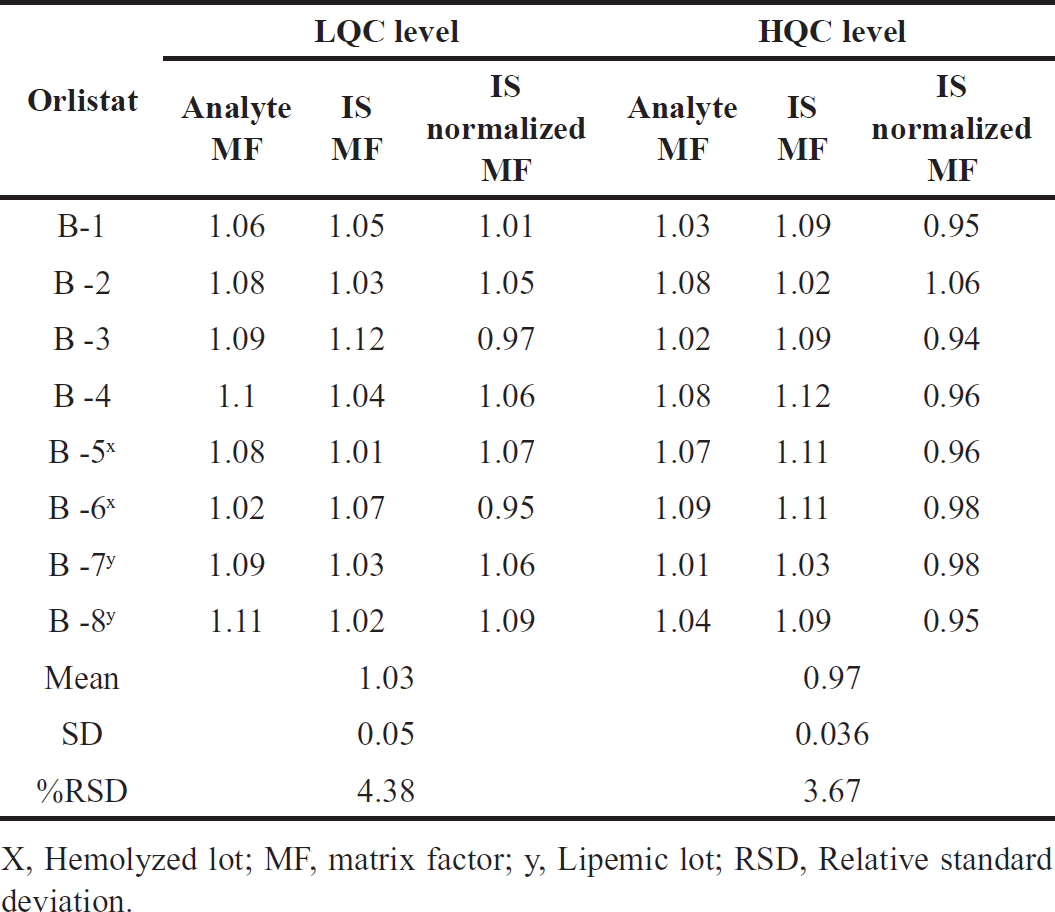 | Table 5. Orlistat findings for matrix effect. [Click here to view] |
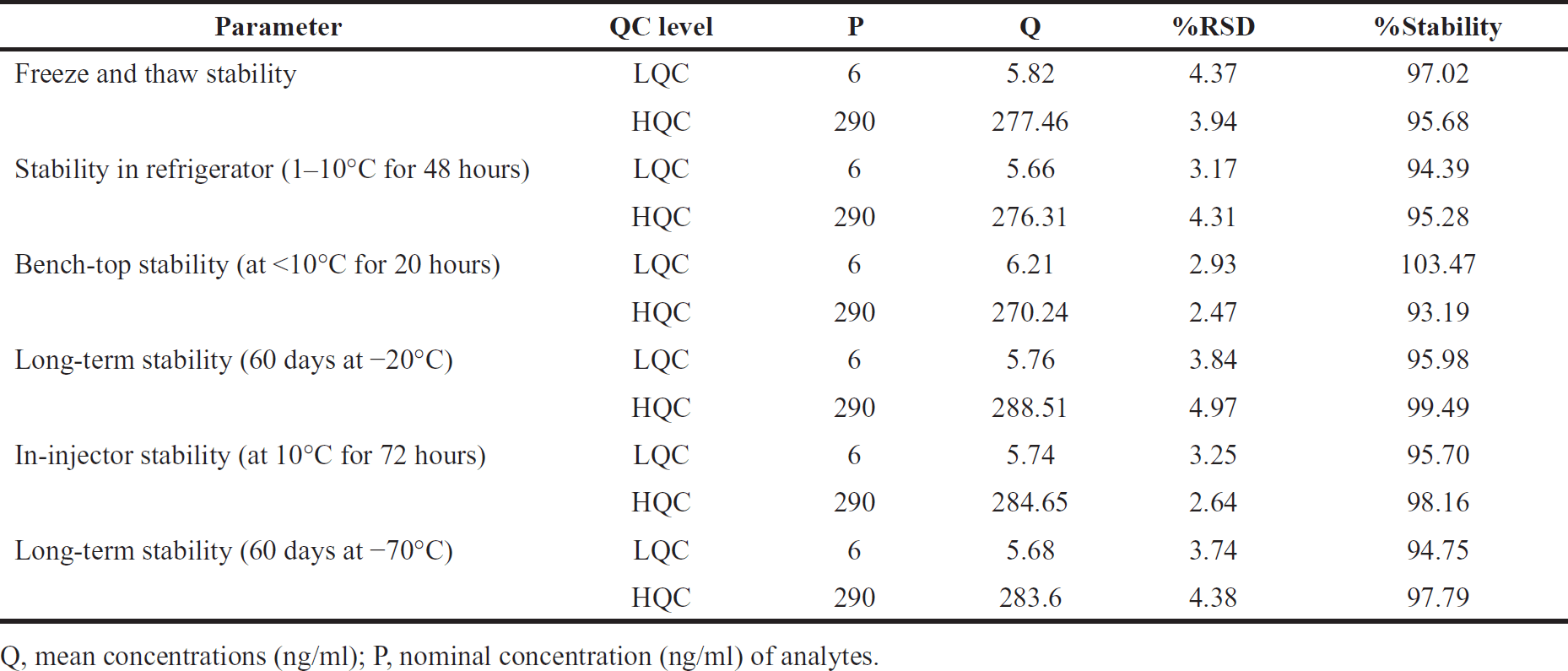 | Table 6. Orlistat stability findings. [Click here to view] |
Stability
Orlistat’s stability was tested in matrix- and aqueous-based samples. Orlistat and IS were unaffected at 1°C–10°C for 70 days and stock solutions in diluent for 48 hours. Matrix stability was tested at −70 and −20 °C for 60 days. The matrix’s stability was tested with a new spiking linear standard. Table 6 displays stability statistics. [12,13,14] After six freeze-thaw cycles below 10°C, the drug sustained for 20 hours. Sample solutions in the auto-sampler lasted 72 hours at 10°C.
Dilution integrity
An experiment was performed to determine Orlistat’s ULOQ (upper limit of quantification) at a concentration twice that of the dilution integrity [13]. Dilution-tested samples had an average back-computed drug content of 85%–115% of the nominal quantity after 1:4 dilutions, with a percent RSD of 4.03.
DISCUSSION
To measure Orlistat in biological matrices, this study set out to develop a sensitive tandem mass spectrometric technique that utilized electrospray ionization and liquid chromatography. Employing a Phenomenex-C18 column and liquid–liquid extraction, the chromatographic elution achieved isocratic separation with a flow rate of 0.80 ml/minute using methanol, acetonitrile, and 0.1% formic acid (65:20:15 V/V/V) as the mobile phase. The selected reaction monitoring revealed parent and product ions at m/z 496.4/142.08 for Orlistat and m/z 501.3/147.07 for Orlistat-D5 IS. The established method exhibited a high linearity (r2 = 0.9999) over the concentration range of 1.2–392.0 ng/ml, with inter- and intra-batch accuracy % relative standard deviation values ranging from 2.84 to 5.32.
The method demonstrated robustness through its exceptional recovery rates of 103.62%, 96.14%, and 95.73% for lower, medium, and higher quality control samples, respectively. The experiment’s DOEs framework, specifically employing a Box–Behnken design, facilitated optimization efforts, with factors such as Capillary Voltage and Cone Voltage significantly impacting Orlistat response (R1). The IS (R2) also showed a substantial correlation with Capillary Voltage and Cone Voltage, with all factors exhibiting statistically significant quadratic effects.
Specificity assessments revealed the absence of matrix material or IS interference, with interfering peaks registering below 20% of the LLOQ samples. The linearity of the method, covering a concentration range of 1.2–392.0 ng/ml, exhibited a high correlation coefficient (r2) of 0.9999. Values of the relative standard deviation for accuracy and precision tests ranged from 2.84% to 5.32%, indicating favorable results both within and between batches. The validated method demonstrated excellent accuracy and precision, which makes it a good fit for clinical and forensic pharmacokinetic and toxicokinetic studies of Orlistat in different biological matrices.
Extraction recovery assessments and matrix effect evaluations further supported the method’s accuracy, with satisfactory results obtained for both Orlistat and the IS. Stability studies under various conditions showcased the method’s robustness over time and different environmental factors. The method’s success is emphasized by its adherence to ICH guidelines and its potential applicability in routine examination of Orlistat in biological samples.
CONCLUSION
In this study, a sensitive and precise LC-MS/MS method was developed and validated to measure Orlistat in human plasma. This method had great specificity, linearity, accuracy, precision, and stability. The linearity equation and correlation coefficient (r2) were y = 0.003546 x – 0.004133 and 0.9999. The developed technique’s intra- and inter-day precision RSD varied from 2.84 to 5.32 percent for QC samples (1.2, 6.0, 196.0, and 290.0 ng/ml). Stability experiments under varied settings showed values between 93.19% and 103.47%. Thus, the validated method can be utilized to study Orlistat’s pharmacokinetics and toxico-kinetics in various biological matrices for clinical and forensic purposes.
AUTHOR CONTRIBUTIONS
All the authors have made substantial contributions to the research work. The primary author is responsible for the conception and design of the study, acquisition of data, analysis and interpretation of data. Other authors have taken part in guiding and supervising throughout the research process, drafting the article, revising it critically for important intellectual content; have agreed to submit to the current journal; gave final approval for the manuscript to be published. All authors agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICJME) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Zhi J, Melia AT, Eggers H, Joly R, Patel IH. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol. 1995;35(11):1103–8.
2. Barbier P, Schneider F. Syntheses of tetrahydrolipstatin and absolute configuration of tetrahydrolipstatin and lipstatin. Helvetica Chimica Acta. 1987;70(1):196–202.
3. Pommier A, Pons M, Kocienski P. The first total synthesis of (−)-lipstatin. J Organ Chem. 1995;60(22):7334–9.
4. Gillies CL, Abrams KR, Lambert PC, Cooper NJ, Sutton AJ, Hsu RT, et al. Pharmacological and lifestyle interventions to prevent or delay type 2 diabetes in people with impaired glucose tolerance: systematic review and meta-analysis. BMJ (Clinical Research Ed.). 2007;334(7588):299.
5. Shekhar Chaudhry, Rajendra B Patil. Stability indicating analytical method development and validation for estimation of orlistat in bulk and its dosage form by HPTLC technique and finding degradants by LC-MS. Am J PharmTech Res. 2018;8(3):131–43.
6. Hitendra Joshi, Yogesh Naliyapara, Vijay Ram, Madhavi Patel, Pragnesh Dave. Quantification of orlistat by a validated, simple, and sensitive high-performance thin layer chromatographic-densitometric assay method. Int J Adv Res Chem Sci. 2017;4(11):23–31.
7. Ray Wieboldt, Dale A Campbell, Jack Henion. Quantitative liquid chromatographic–tandem mass spectrometric determination of orlistat in plasma with a quadrupole ion trap. J Chromatogr B. 1998;708:121–9.
8. Sreekanth Nama, Babu Rao Chandu, Mukkanti Khaggaet. A new RP-HPLC method development and validation of orlistat in bulk and pharmaceutical dosage forms. Int J Pharm Sci Res. 2010;1(6):251–7.
9. Xiao Song, Zhu Xiao-Lan, Chen Bo, Yao Shou-zhuo. Determination of orlistat in capsules by HPLC/MS. Chinese J Pharm Anal. 2005;25(9):1055–7.
10. European Medicines Agency, Guideline on bioanalytical method validation 2011. FDA guidance for industry, bioanalytical method validation, US Department of Health and Human Services, Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM) May 2001.
11. Henion J, Brewer E, and Rule G. Sample preparation for LC/MS/MS: knowing the basic requirements and the big picture of an LC/MS system can ensure success in most instances. Anal Chem. 1998;70:650A–6A.
12. Chambers EE, Woodcock MJ, Wheaton JP. Systematic development of a UPLC–MS/MS method for the determination of tricyclic antidepressants in human urine.J Pharm Biomed Anal. 2014;88:660–5.
13. Patel DS, Sharma N, Patel MC. Development and validation of a selective and sensitive LC–M/MS method for determination of cycloserine in human plasma: application to bioequivalence study, J Chromatogr B. 2011;879:2265–73.
14. Hindu K, Vinodhini C, Srinivas SK, Rajan SM, Chitra K, Mangathayaru K. Validated RP-HPLC method for quantification of paclitaxel in human plasma—eliminates negative influence of cremophor EI. IJCRR. 2018;10(13):5–10.
15. Murphy AT, Kasper SC, Gillespie TA, DeLong AF. Determination of xanomeline and active metabolite, N-Desmethylxanomeline, in human plasma by liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J Chromatogr B Biomed Appl. 1995;668:273–80.
16. Shankar CH, Bhikshapathi DVRN. Validation of a specific LC-MS/MS method for the quantification of anti-cancer alectinib in biological matrices. IJPR. 2021;13(1):6513–21.
17. Bodkin J, Humphries E, McLeod M. The total synthesis of (−)-tetrahydrolipstatin. Aus J Chem. 2003;56(8):795–803.
18. Johns BA, Kawasuji T, Weatherhead JG, Taishi T, Temelkoff DP, Yoshida H, et al. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem. 2013;56(14):5901–16.
19. Sanlialp Zeyrek A, Takmak S, Kurban NK, Arslan S. Systematic review and meta-analysis: physical-procedural interventions used to reduce pain during intramuscular injections in adults. J Adv Nurs. 2019;75(12):3346–61.
20. Sisson H. Aspirating during the intramuscular injection procedure: a systematic literature review. J Clin Nurs. 2015;24(18):2368–75.
21. Spreen W, Min S, Ford SL, Chen S, Lou Y, Bomar M, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203.