
INTRODUCTION
Generic medicines are the most affordable source of essential medicines in the continuously changing pharmaceutical industry. The Hatch Waxman Act of 1984 encouraged the worldwide development of generic drugs [1]. As the growth of generic companies rapidly increases, innovator companies focus on complex drug discovery and development. In recent years, in 2016, a particular group of generics called complex generics has become a more challenging point for generic pharmaceutical companies [2]. Unlike simple generics, complex generics pose unique challenges in their developmental stage, manufacturing stage, regulatory approval process, and use. These generics are also known by different names such as specialty generics, super generics, hybrid drugs, value-added generics, and off-patent medications. The complex generic market is estimated to be 84 US billion by 2024 and has anticipated to grow at a CAGR of 8% within the coming 10 years [3].
What are complex generics?
The term “complex generic” is legally acknowledged only in the US and, according to the US Food and Drug Administration (FDA), complex generics are generic versions of complex drugs [4]. Complex drugs are drugs that contain either a non-biological or biologically active ingredient. Most of these complex generics are non-biologics and are called non-biological complex drugs (NBCDs). NBCDs have large heteromolecular structures and are difficult to isolate, quantify, and fully characterize using physicochemical analytical methods [5]. Only a few biologics have been approved by the US FDA as complex generic drugs. The most common are enoxaparin sodium injections, glatiramer acetate, heparin, and low-molecular-weight heparin molecules. Sanofi-Aventis manufactured and marketed enoxaparin under the brand name Lovenox and as a generic drug under the name enoxaparin sodium for injection (30 mg/0.3 ml and 40 mg/0.4 ml prefilled syringes; 60 mg/0.6 ml, 80 mg/0.8 ml, and 100 mg/1ml graduated prefilled syringes; 300 mg/3 ml Multiple-dose vial) manufactured by Winthrop US (a Sanofi company) [6]. Two generic Lovenox versions have been approved by the FDA [7]. To approve a complex natural drug, generic companies need to demonstrate equivalence in fundamental reaction schemes, physiochemical characterization, and process signatures. In the case of complex biological generic approval, data relating to the similarity of biochemical/biological markers serve as a confirmatory test [8].
As per the US FDA, Generic Drug User Fee (GUDFA) Amendment II commitment letter, “Complex generics are those drug products generally include products with complex active ingredients, complex formulations, complex routes of delivery, complex dosage forms OR complex drug-device combination products OR other products where complexity or uncertainty concerning the approval pathway or possible alternative approach would benefit from early scientific engagement” [9]. As per the US FDA Orange book “Drug Products are considered to be therapeutic equivalents only if they are pharmaceutical equivalents and if they can be expected to have the same clinical effect and safety profile when administered to patients under the conditions specified in the labeling”[10]. According to the European Medicines Agency (EMA), these complex generics are termed “hybrid medicines”. EMA refers to “hybrid medicines are medicines whose authorization depends partly on the results of tests on the reference medicine and partly on new data from clinical trials” [11].
Classification of complex generics
Although classification is needed for overall decision-making, there is no specific classification for complex generics worldwide. Therefore, the US Center for Drug Evaluation and Research (CDER) roughly classifies these drugs based on different types of complexity [12]. This rough classification is mentioned below in Table 1 [12,13].
Encouraging the development of complex generics is critical for ensuring patient access to a diverse range of potentially cost-saving treatments. The FDA envisions collaboration with industry as an opportunity to facilitate complex generic development, evaluation, approval, and market authorization [14]. However, concerning complex generic drugs, concerns have arisen regarding the limited competition and paragraph IV certification filed by generic manufacturers, potentially holding up the entry of complex generic drugs into the global market [15]. By considering all these challenges, this study explores the regulatory challenges that reduce the market approval of these products. Moving deeper into the current regulatory framework highlights the main manufacturing hurdles and the steps taken to overcome them. Innovative solutions are still being developed to ensure the quality and efficacy of complex generics [2]. Based on some case studies, an overview of different classes, their challenges, and the successful steps taken are also highlighted in this paper. Although a complex generic represents the upcoming future of generic industries, the reasons behind their slower development are still the most significant hidden fact. This study comprehensively overviews some major challenges of different complex generic products. Ultimately, this review offers insightful information to all stakeholders for the development of high-quality and affordable complex generic products.
RECENT TRENDS IN COMPLEX GENERICS
As the demand for cost-effective alternatives to complex drugs grows globally, complex generics have become a focus for domestic and foreign pharmaceutical companies. Despite an estimated 91% of all prescriptions in the United States being filled as generic drugs, the approval and adoption of different classes of complex generics have lagged, mainly because of difficulties in bringing them to the market [16]. The critical development in complex generics is establishing the Center for Research on Complex Generics (CRCG) by the US FDA. CRCG aims to enhance the collaboration of generic companies to combine resources and expertise, increasing investment in research and development (R and D), manufacturing, and marketing of complex generics. The CRCG supports all stakeholders by conducting collaborative research, training and webinars, workshops, focal group discussions, scholarly project presentations, and other initiatives [17]. The pre-ANDA program was included in the Generic Drug User Fee Act (GDUFA) II of the US FDA to provide early engagement of generic manufacturers with regulatory agencies to promote a more effective and efficient review process [18]. Product-specific guidelines are one of the significant upcoming ways to grow certain complex generic drugs rapidly [19]. In May 2023, the FDA published 47 product-specific guidelines (PSGs) drafts, of which 25 were for complex generics [14]. Biosimilars are considered a subcategory of complex generics because of their complex developmental and evaluation processes. Technological advancements have played an essential role in developing complex versions of these drugs. A recent review demonstrated methodological advancements, such as the use of nuclear magnetic resonance spectroscopy, high-resolution mass spectrometry, and multivariate statistical analysis, for structural studies of natural complex drugs, such as conjugated estrogens and glatiramer acetate [8]. The FDA GUDFA Science and Research Program insists that generic industries focus on quantitative methods and modeling to establish bioequivalence [20]. The regulatory cheerful lights have taken up the above advancement on the market dynamics of complex generics. In February 2022, US FDA approved the first generic version of complex injectable apokyn (apomorphoine hydrochloride pen injection) manufactured by Sage Chemicals [21]. In 2022, the FDA authorized the first generic versions of Restasis (cyclosporine ophthalmic emulsion) and Symbicort (budesonide + formoterol fumarate dehydrate inhaler) by Mylan Pharmaceuticals [22]. The first generic Vivitrol (naltrexone for extended-release injectable solution) of Teva Pharmaceuticals and Spiriva HandiHaler (Tiotropium Bromide) of Lupin Inc were authorized by the FDA on 2023 [23]. Current research in complex generics highlights that most researchers in developed and developing countries are now focusing on complex generics. Papers relating to complex generics were retrieved from Scopus on November 11, 2023, using the “complex generics.” A total of 120 articles were exported, and the conclusion drawn was the number of publications is increasing dramatically whereas the majority of the papers belong to regulated markets such as the US and European countries, as depicted in Figures 1. From these studies, it can be concluded that high-quality pharmacoeconomic studies, modeling approaches, and standardized regulatory guidelines are required for the future development of these complex generics.
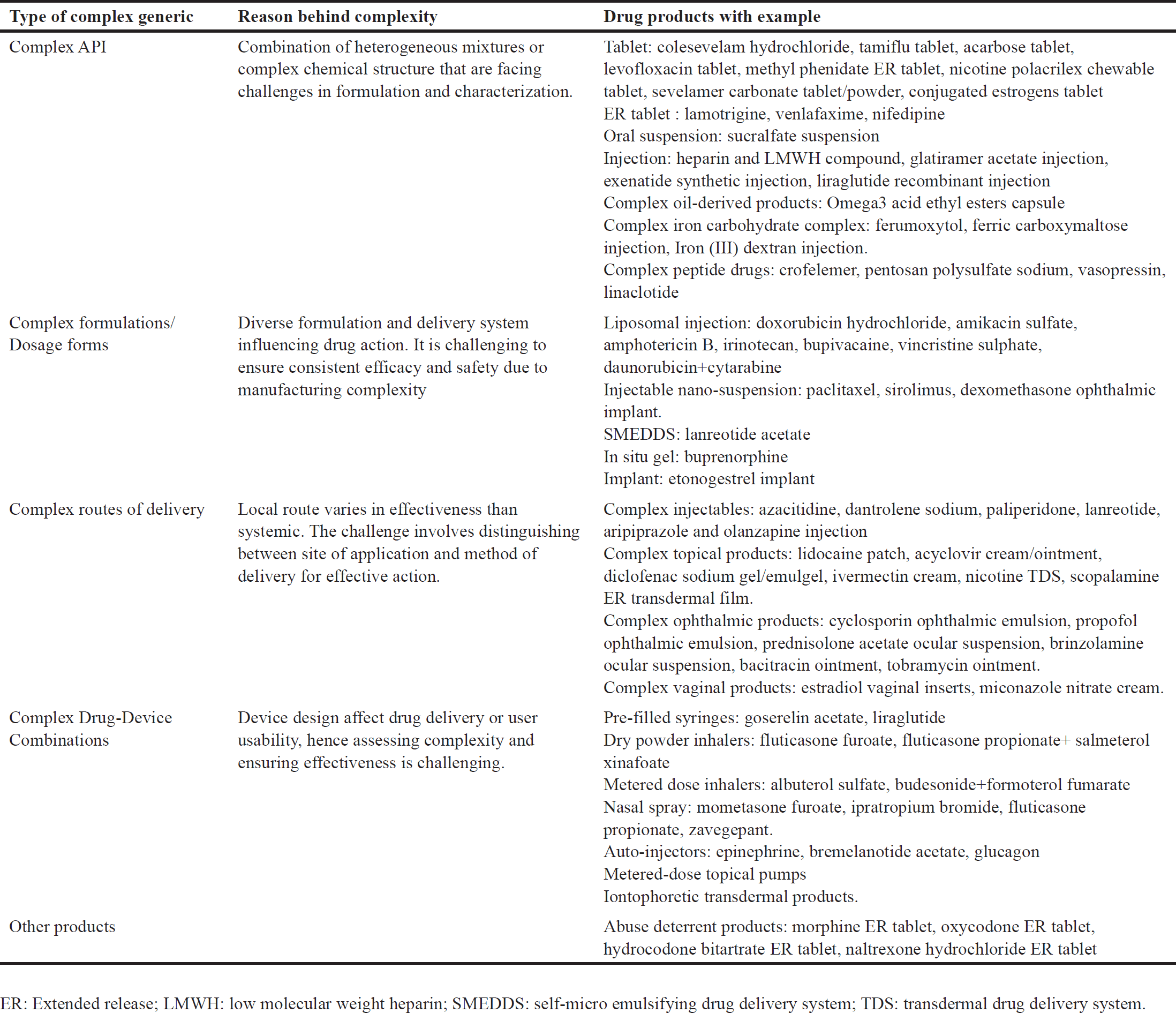 | Table 1. Classification with examples of drugs under each class of complex generics. [Click here to view] |
CHALLENGES OF COMPLEX GENERICS
The landscape of complex generics is evolving rapidly with technological advancements to overcome its multifaceted challenges. The journey to understand the challenges associated with complex generics is fraught with more complex obstacles in each stage of its lifecycle (starting from quantification, manufacturing, characterization, clinical trials, regulatory approval, market dynamics, and so on). The main challenges in the manufacturing and characterization stages of complex generics are lack of expertise and equipment, use of more complex mixtures of components or excipients, lack of analytical methodologies, inappropriate tracing of the exact delivery route, and difficulty in proving in vitro bioequivalence.
Technical challenges are more common in the case of complex injectables, dermal products, and drug-device combination products. USP conducted an open forum survey and concluded that in vitro dissolution techniques, physicochemical characterization, complex excipient monographs, and their analysis were the most common developmental challenges associated with complex injectables [24]. Compared to traditional generics, a more sophisticated planning and development process is required to overcome the regulatory challenges of complex generics. Due to a lack of proper regulatory guidance, additional comparative characterization and clinical study data must be submitted depending on various regulatory authority requirements for the same product [25]. Due to the impact of direct and indirect production costs, it is clear that regulatory and developmental challenges are closely related. In turn, these challenges result in lower market access and higher prices. Regulatory agencies have taken several steps to overcome these challenges. Draft guidance for pre-ANDA meetings [26], guidance to determine the suitable time to submit ANDAs for certain complex generics (peptides), workshops focusing on sophisticated quantitative and computational modeling, and providing product-specific guidance are some of the steps adopted by the US FDA to overcome these challenges [27]. A study by Stern et al. [27] highlighted the importance of advanced research and education by emphasizing complexities. This study underscores the significance of collaboration between industry, academia, and regulatory agencies in developing standard guidelines [28]. Understanding these factors influencing the demand and supply of medicines can help policymakers and healthcare providers increase the supply and use of these affordable medicines [29].
Challenges relating to non-biologic complex generics
NBCDs are challenging to duplicate owing to their complex nature and regulatory uncertainties. Examples include glatiramer, iron-carbohydrate complexes, polymeric micelles, complex ocular emulsions, parenteral microspheres, liposomes, injectables, implantables, and transdermal and locally acting products [30]. Complex molecular structures and manufacturing processes contribute to their unique pharmacokinetic and pharmacodynamic properties. Thus, it is challenging to demonstrate therapeutic equivalence using traditional bioequivalence tests [31]. In other circumstances, bioequivalence may not be sufficient to assess therapeutic efficacy. NBCDs are not approved through a centralized approach; instead, their safety and efficacy are determined on a case-by-case basis, and in some rare circumstances, an approach similar to biosimilars is used [32]. The EMA has issued reflection papers on nanomedicine products, such as liposomal systems, iron-core nanoparticles, micellar systems, and coated nanosystems, but it has yet to review all NBCDs systematically [33]. These papers reflect the ongoing challenges in this class of generics. Doxorubicin hydrochloride liposomal injection is a successful NBCG with complex manufacturing challenges (requiring 17 different process vessels, specialized equipment, and a time-consuming, complex process) [31]. The development of robust analytical methods can ensure batch-to-batch consistency; however, it is also a challenging factor when considering the cost of manufacturing. The successful development of glatiramer acetate injection, sevelamer carbonate tablet, iron sucrose injection, and iron dextrose injection highlights the potential need for a comprehensive regulatory guideline that contains comprehensive characterization techniques and additional clinical data [34]. Lygature, a public-private partnership pioneer in 2009, started the NBCD working group for discussions among various stakeholders, thus improving the safety and efficacy of NBCD drugs and their follow-on products [29]. The challenges based on different classes of NBCD are discussed in detail in Table 2.
Challenges relating biologic complex generics
Developing biological substitutes based solely on BE and pharmaceutical equivalence (PE) assessments, such as generic NBCDs, is impossible. In addition, because of the heterogeneity of both API and excipients and the challenges in implementing sensitive analytical techniques, identifying these components is inconvenient and indirectly affects PE assessment [31,35]. However, differences in protein structure might impact pharmacodynamics without changing bio-distribution; therefore, an identical profile alone does not guarantee therapeutic equivalency [36]. Given the complexity of protein molecules and the limitations of current analytical methods, it may be challenging for manufacturers to demonstrate the sameness in an active ingredient of the generic version with that of RLD. Thus, it is clear that ANDAs are not a focus 505(j)(2)(A) of the FD and C Act guidance [37].
Similar to generics, the demonstration of bioequivalence is insufficient to prove the similarity of biological/naturally derived complex drugs [38]. Regarding generics, all three complete CTD modules are available, with the additional stimulation that Module 3 must include data from the comparability experiment. In contrast to generics, biosimilars should, when applicable, be included in modules 4 and 5, together with the findings of non-clinical and clinical comparability studies. In 2009, the FDA employed a comparable study strategy to establish a shortened authorization process for biosimilars under the BPCI Act [37].
Applicants must specifically show the equivalency of the following: (i) the mode of de-polymerization and the source material of heparin; (ii) determination of physicochemical properties using particular analytical techniques (both API and excipients); (iii) the sequence of oligosaccharide molecules, fragment mapping, and disaccharide building blocks; (iv)biological assays and bioassays; and (v) in vivo pharmacodynamic studies [39].
Falconer et al. [40] described the theoretical and operational factors that should be considered when choosing characteristics and test techniques for biosimilars, evaluating potential, and assigning analytical measures to the U.S. FDA analytical similarity evaluation. The methods used to identify and define CQAs for biologics may be examples for developing sophisticated nanomedicine medicinal products [42]. Regarding the regulatory aspects of complex natural drugs, even different regulated markets have different approval processes. For example, low molecular weight heparins were approved as the generic version of non-biologics by the US FDA, whereas the same was approved via a biosimilar approach in EMA [31]. Many product and process- and process-related challenges are also associated with biologics, including choice of study population, high inter- and intra-subject variability, change in parameters with disease progression, and difficulty in interpretation [53].
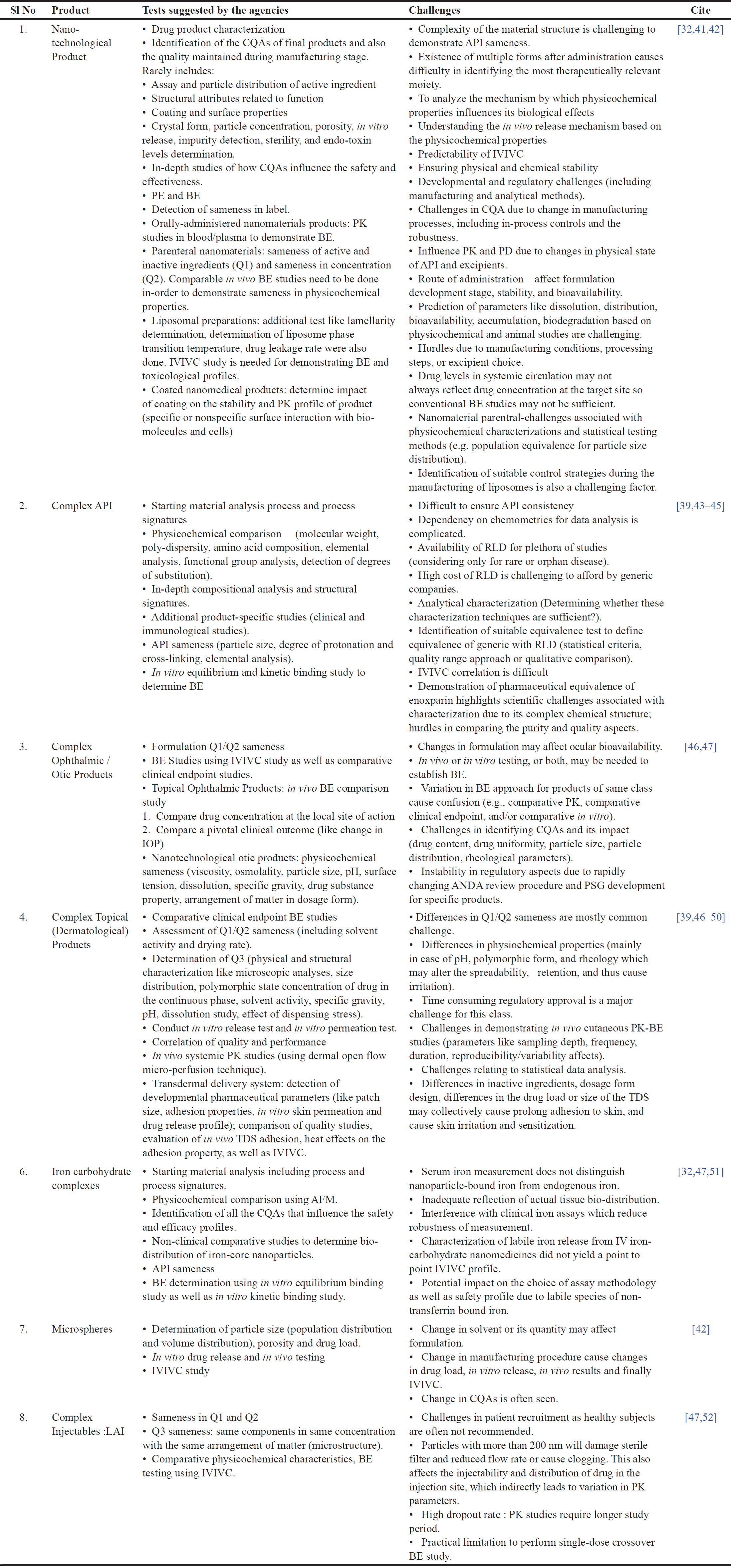 | Table 2. Challenges based on different classes of NBCDs. [Click here to view] |
Challenges relating to drug-device combination complex generics
There is a growing assimilation of drug-device combination products owing to several regulatory complications. Sanduria et al. [54] highlight that countries such as the US and EU have vast differences in regulatory approval pathways, and countries such as China, Japan, and India need more well-developed guidelines. The steps in registering a generic, orally inhaled drug product in a particular market may differ depending on the country. These steps include comparing the similarity of device performance and formulation to that of the original product and conducting tests to compare the product’s in vitro and in vivo aspects [55].
Donnelly et al. [56] thoroughly analyzed complex medicinal products supplied via the female reproductive tract and highlighted the difficulties encountered in creating generic medicines due to a lack of adequate bioequivalence techniques for locally acting pharmaceuticals. The authors emphasize FDA-funded research collaboration with the University of Buffalo (New York) to create an open-source, generalized physiologically based pharmacokinetic (PBPK) modeling and simulation platform for complex drugs administered through the female reproductive tract [56]. Sharan et al. highlight obstacles to developing generic intrauterine system drug products [56]. All of this research indicates that there are difficulties in evaluating combination products, as well as deficiencies in regulatory guidelines [57]. Combination products need help with the methods available for testing, developmental and characterization techniques, product standards as per regulatory specifications, and so on. Some common challenges of drug-device combination products are below [58–61].
a. Lack of expertise and skilled personnel.
b. In-house testing is challenging due to the lack of quality system registration, whereas testing outside needs more investments.
c. Lack of industrial guidance results in considering the device of one country as a drug in another country.
d. Variation in BE result due to device design, complex PK assessment due to the local action, and potential discrepancies in in vitro-in vivo correlation (IVIVC).
e. Use of lengthy comparative clinical endpoint studies as suggested by some regulatory agencies.
f. Challenges in using sensitive In-vitro BE assessment techniques such as particle size determination, device performance, and aerosol characterization. g. Ensuring both formulation sameness and device similarity is the most challenging.
Other challenges associated with complex generics
Apart from developmental, manufacturing, technical, and regulatory challenges, complex generics are associated with other challenges, such as economic, market, patient, and industry-related.
Economic challenges
Manufacturing therapeutic equivalent duplicates is a viable strategy for lowering medical expenses and increasing the financial stability of manufacturing companies. Pharmacoeconomics suggests that excessive research and developmental expenses can have two effects: either a low return on investment (which could result in a product being removed from the market) or an excessively high price (which would render the medication unavailable to the majority of patients) [62].
Market challenges
Companies are more likely to manufacture a profitable generic version of a branded drug with a high average wholesale price [1]. The challenge in introducing complex generics is pinpointing target markets to fulfill unmet patient needs. Another problematic element in the market is the competition for patients. One of the biggest challenges is combating anti-generic initiatives purportedly supported by associations between physicians and patients [63].
Patient and industry challenges
These challenges mainly involve those associated with planning, study design, and site selection. Biopharmaceutical businesses must understand the patient population needed in their trials to secure approval in their intended markets. They should consider how to handle the risk of attrition and non-compliance among small patient populations. Many investigators may not be interested in generic medication trials because of a lack of scientific or medical interest in non-novel drug studies or because they may not be sufficiently motivated to participate in a generic trial. Thus, biopharmaceutical businesses require a partner who can recognize sites with a sufficient patient pool and is eager to provide access to generic options through trial participation. All these above criteria become challenging factors for both industry and patients in terms of investment and compliance challenges [64].
Challenges associated with the development of an analytical tool
Because of its complexity, developers find it difficult to establish therapeutic equivalence (PE+BE) and identify and characterize CQAs in the developmental stages of complex generics. Recognizing the current limitations, stakeholders now focus on additional research to clarify the mechanisms of action and develop and validate relevant analytical tools [63]. The deployment of such advanced technologies should be considered product-by-product, which can influence the safety and efficiency profile. Zhang and Lionberger [65]highlighted traditional quantitative mathematical modeling tools, such as exposure-response modeling, population pharmacokinetic analysis, PK-PD/PBPK modeling, and clinical trial simulation, which have made significant impacts and are becoming indispensable in the development and review of complex generic drugs, which are expected to reduce the time and cost of production [65,66]. Troiano et al. [67] demonstrated how they employed a quality-by-design strategy to develop a generic version of complex nanomedicines. Specifically, they created an in-depth awareness of the products, processes, and technology used by applying a risk-based methodology to discover and classify the product features and process factors. Although this strategy primarily focuses on chemistry, manufacturing, and control issues, drug development’s preclinical, clinical, and regulatory elements are also carefully considered [67]. Despite model-informed drug development being included in Section 3 of the Prescription Drug User Fee Act, many pharmaceutical companies must utilize or be aware of it. Model-informed drug development (MIDD) applications for GDUFA raise similar concerns. As many smaller companies are involved, transforming MIDD from a luxurious method to an essential method could be a significant issue for generic manufacturers [68]. Furthermore, the lack of a scientific understanding of the IVIVC of complex pharmaceuticals, combined with characterization challenges, influences API comparison studies and the in vitro profile, hindering the development of generic lower-cost complex drugs.
REGULATORY SCENARIO OF COMPLEX GENERICS IN DIFFERENT COUNTRIES
To ensure therapeutic equivalence and minimize the need for extensive clinical trials, regulatory agencies worldwide have established guidelines and requirements for generic manufacturers. However, developing and approving generic versions of complex products necessitate additional studies beyond the scope of general guidelines for simpler generics. The current regulatory status in different countries is outlined below and summarized in Figure 2.
Europe
Under Directive 2001/83/EC.19, the European Union regulates pharmaceutical items for human use [69]. Marketers must submit an ANDA via a centralized, decentralized, or mutual recognition procedure to obtain market authorization for hybrid drugs. Additionally, pre-clinical or crossover comparative clinical studies, along with data from 505(b) [2] applications, are adopted in the case of European countries. The hybrid application method based on Article 10 [3] also examines the safety of complex generics compared to RLD [70].
Japan
In Japan, drugs are assessed by the Pharmaceuticals and Medical Devices Agency (PMDA) and approved by the Ministry of Health, Labour, and Welfare. The PMDA Office of generic drugs provides multiple consultations to generic manufacturers to encourage safe, efficient, and affordable drugs in a consistent supply to the public. “Consultations on BE and the quality of generic drugs” of 2011 highlights that blood concentration does not serve as a BE index for specific formulations such as inhalers and nasal and ophthalmic products. Therefore, pharmacodynamic or clinical endpoint BE research is considered to approve such generics [71].
Canada
Health Canada approves complex generics through generic, biosimilar, or new drug application (NDA) pathways. The approval procedure varies on a case-by-case basis and follows the pre-ANDA submission procedure. ANDA applications for generic versions of certain pharmaceuticals have received a notice of non-compliance or deficiency [69]. According to the Biologics and Genetic Therapies Directorate, pharmaceutical goods containing LMWHs are claimed to be similar to another heparin already on the market. These data show that Canada treats NBCDs as complex generics and includes biologics [72].
Brazil
The Brazilian Regulatory Authority Agencia Nacional de Vigilancia Sanitaria published RDC No.60/2014 for the market authorization of all types of generic drugs [73]. The approval of complex generics depends on this procedure and LatAm (Latin American countries) guidelines. Glatiramer acetate generics was first approved by LatAm based on US FDA guidelines, and later approved using the policy based on guidelines for small-molecule chemical synthesis or even less stringent specifications (for example, bioequivalence studies were not required). The change in the use of guidelines summarizes and emphasizes the importance of clear and harmonized regulatory procedures and appropriate guidelines for ensuring the quality and safety of complex generics or follow-on medicines [70].
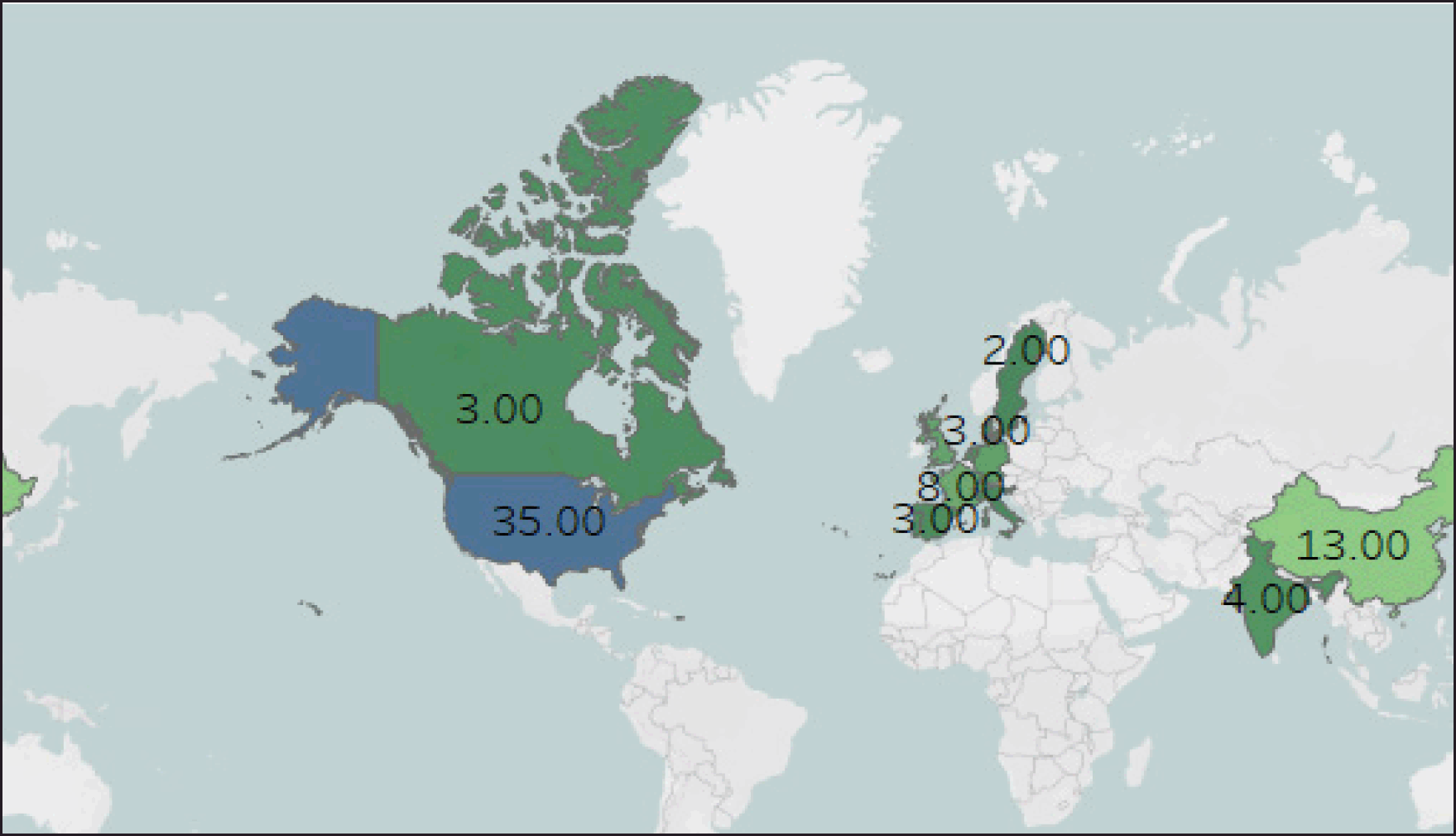 | Figure 1. Number of publication per country wise in Scopus database (Using Tableau Public software). [Click here to view] |
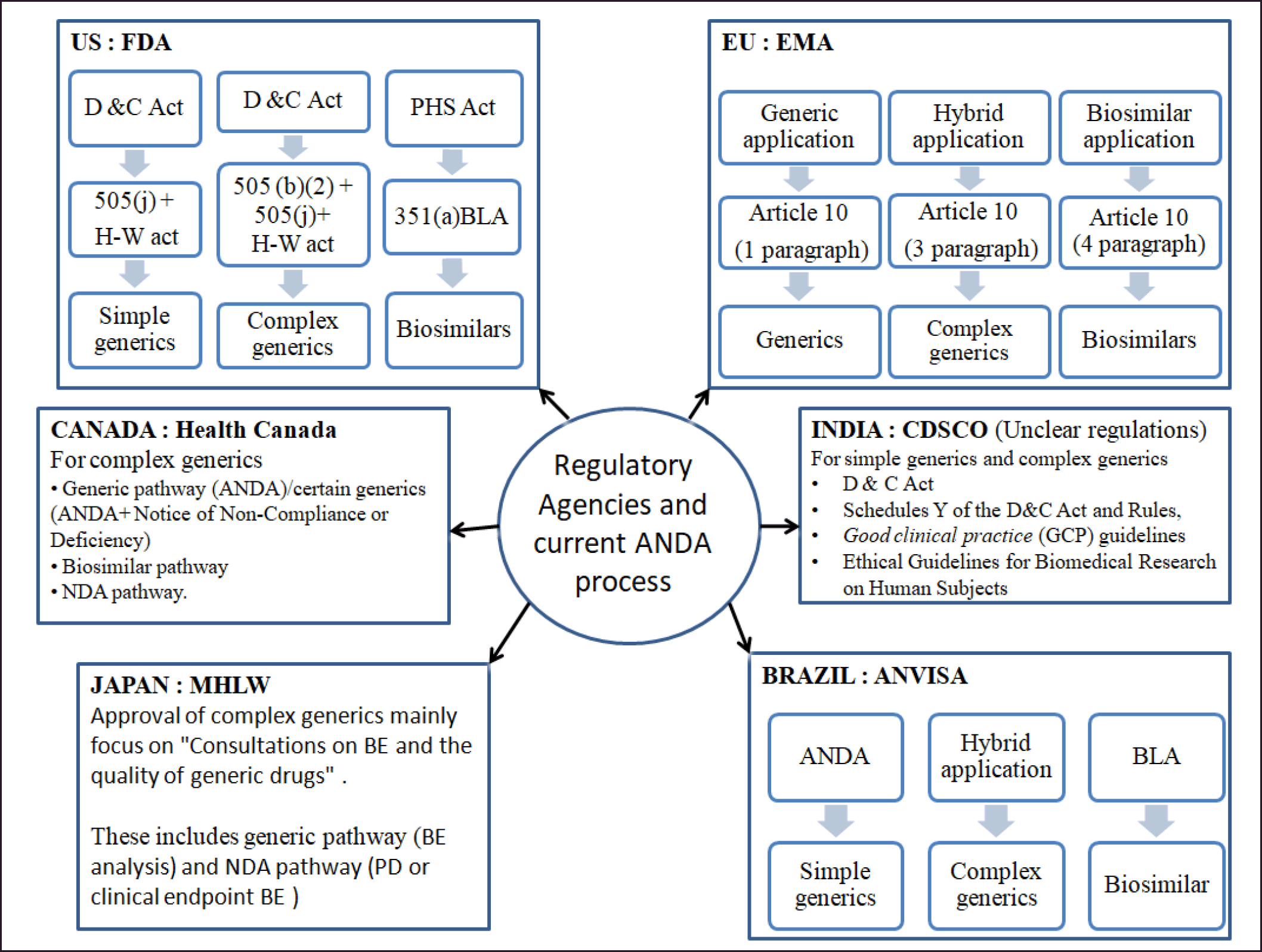 | Figure 2. Current regulatory status for complex generics in different countries. [Click here to view] |
United States
The growth of generics advanced with the emergence of the GDUFA. GDUFA I aim to efficiently evaluate the equivalence of the generic version of the drug to that of RLD. GUDFA II mainly brings the pre-ANDA program and controlled correspondence. FDA officials and ANDA applicants collaborated in the pre-ANDA program to address regulatory uncertainty and reduce review cycles. It also highlights the need for PSG and starts issuing it after this program [67]. The US FDA has issued new and revised product-specific guidance for the most challenging products to aid in developing, manufacturing, and regulatory approval of their generic version, which is considered safe and effective. The guidance document “Determining Whether to Submit an ANDA or a 505(b) (2) Application—Guidance for Industry” concluded that most of the complex generics use the 505(b)(2) pathway for getting regulatory approval in the US [74]. Complex generics can be reviewed via a 505(j) application (complex dosage form/formulation as well as complex route of delivery) and 505(b)(2) approval pathway (most likely complex API).
India
The Drugs Controller General of India has yet to establish precise criteria for evaluating the efficacy and safety of complex generics. The Central Drugs Standard Control Organization (CDSCO) Guidelines for bioavailability and bioequivalence studies must be followed when submitting generic drug applications [75]. Guidelines/rules such as Rule 122 A to E of the Drugs and Cosmetics Act (D and C Act), Schedules Y of the D and C Act and Rules, Good Clinical Practice [GCP] guidelines released by CDSCO, and Ethical Guidelines for Biomedical Research on Human Subjects govern all trials in India; hence, establishing the regulatory procedure for registering a second-entry product (even though it is a complex generic/ simple generic) considers all these guidelines [56]. Unlike in other countries in India, the regulation of combination products is unclear; some products in this category, such as drug-eluting stents, are considered drugs regardless of the device component linked to them [59].
FUTURE ADVANCEMENT IN THE FIELD OF COMPLEX GENERICS
Future studies should focus on overcoming these challenges and developing standardized procedures for assessing and approving complex generics. Harmonizing international regulatory guidelines would ensure patient access to safe and effective generic pharmaceuticals while facilitating manufacturers to access the market. Furthermore, regulatory frameworks must be continuously monitored and adjusted to keep pace with the developments in pharmaceutical technology, including nanomedicine products. Stakeholders can collaborate to eliminate regulatory obstacles and improve the availability of high-quality complex generics to meet the requirements of patients worldwide by encouraging cooperation and innovation. Analytical advancements, modeling, and simulation are some of the scientific achievements related to generic medications. Analytical advancements, such as in vitro characterization technologies can be used to characterize complex API structures, evaluate formulation CQAs, and also to determine bioequivalence. This can thereby reduce the need for comparative clinical endpoint BE studies of various formulations.
The FDA proposed “Further Opportunities for Harmonization of Standards for Generic Drugs,” which was supported by ICH to develop globally harmonized guidelines for generic drugs (including complex generics). Harmonization of the theoretical, scientific, and technical aspects of complex generics can significantly improve public health by accelerating the supply and increasing patient access globally.
CONCLUSION
The regulatory framework for generics in various countries is complex and involves multiple approaches and challenges. Regulatory bodies in the US and Europe have established procedures for approving complex generics; however, their requirements and methods vary. A recurring issue is understanding the unique challenges presented by complex generics, which often require further research in addition to conventional bioequivalence evaluations. Physicochemical characterization, in vivo bioequivalence testing, and occasional clinical investigations are needed to prove product safety and efficacy. Complex generics face several challenges, including variable physicochemical characterization, difficulties demonstrating equivalency, managing patent concerns, and commercial exclusivity. Furthermore, because there are no precise regulatory requirements, research is expensive. Thus, firms are incentivized to look into new methods for regulatory approval. Furthermore, there are discrepancies in the approval processes for similar items due to the need for harmonization between regulatory criteria in different countries. Notwithstanding these obstacles, business is expected to grow considerably in the upcoming years because of the growing need for affordable medications. Complex generic development is the current trend, and adjusting to more high-quality development procedures will help achieve success. To accomplish significant “time-to-market” strategic and operational goals, pharmaceutical companies should follow the best practices for regulations regarding where, what, when, and how to comply. To develop harmonized regulatory standards, there should be collaboration between businesses, academia, and regulatory bodies.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Lee CY, Chen X, Romanelli RJ, Segal JB. Forces influencing generic drug development in the United States: a narrative review. J Pharm Policy Pract. 2016 Dec;9(1):26.
2. Lionberger R. Generics Drugs in the 21st Century: FDA’s Actions Create Transparency and Value for Complex Generic Product Development.2021. [cited 2024 Jan 8]. Available from: https://www.fda.gov/drugs/news-events-human-drugs/generic-drugs-21st-century-fdas-actions-create-transparency-and-value-complex-generic-product.
3. Super Generics Market Size and Share Revenue Growth 2035 [Internet]. [cited 2024 Jan 8]. Available from: https://www.rootsanalysis.com/reports/super-generics-market/275.html
4. GDUFA Reauthorization Performance Goals and Program Enhancements Fiscal Years 2023-2027[Internet]. [cited 2024 Jan 8]. Available from: https://www.fda.gov/media/153631/download
5. Schellekens H, Stegemann S, Weinstein V, De Vlieger JSB, Fluhmann B, Mühlebach S, et al. How to regulate non-biological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014 Jan;16(1):15–21.
6. Lovenox® for Anticoagulant Therapy. Sanofi [Internet]. [cited 2024 Jun 26]. Available from: https://www.lovenox.com/dosing-and-administration
7. Research C for DE and. Generic Enoxaparin Questions and Answers. FDA [Internet]. 2018 Nov 3 [cited 2024 Jun 26]. Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/generic-enoxaparin-questions-and-answers
8. Sasisekharan R, Lee SL, Rosenberg A, Walker LA, editors. The Science and Regulations of Naturally Derived Complex Drugs [Internet]. Springer International Publishing, Cham, Switzerland, 2019 [cited 2023 Oct 13]. Available from: http://link.springer.com/10.1007/978-3-030-11751-1
9. GDUFA II Commitment Letter 5/12/16, GDUFA Reauthorization Performance Goals and Program Enhancements Fiscal Years 2023-2027[Internet]. [cited 2024 Jan 8]. Available from: https://www.fda.gov/media/101052/download
10. Research Center for Drug Evaluation. Approved drug products with therapeutic equivalence evaluations| Orange Book. Silver Spring, MD: FDA [Internet]; 2023 Oct 13 [cited 2023 Oct 17]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book
11. EMA. European Medicines Agency. Generic and hybrid medicines. 2018 [cited 2023 Jan 10]; Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/generic-hybrid-medicines.
12. Jiang W, Kozak D, Zhang D, Wang Y, Raney S G, Zhang Y, et al. How are complex drug products defined and classified? office of research and standards, office of generic drugs, CDER, FDA [Internet]. 2019 Apr [cited 2023 Jan 1]; Available from: https://pqri.org/wp-content/uploads/2019/04/Abstract-Complex-Drug-Product-Classification-Criteria-updated.pdf
13. Manual of policies and procedures. CDER.FDA [Internet].[Cited on 2023 Oct11]. Available from: https://www.fda.gov/media/157675/download
14. Research C for DE and. The Center for Research on Complex Generics. FDA [Internet]. 2023 Apr 5 [cited 2023 Jan 1]; Available from: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/center-research-complex-generics
15. Center for Drug Evaluation and Research Fiscal Year 2022. GDUFA Science and Research Report. [cited 2023 April 2]. Available from: https://www.fda.gov/media/ 16484 3/download.
16. Research C for DE and. Office of Generic Drugs 2022 Annual Report. FDA [Internet]. 2023 Mar 1 [cited 2023 Oct 13]; Available from: https://www.fda.gov/drugs/generic-drugs/office-generic-drugs-2022-annual-report
17. Research C for DE and. The Center for Research on Complex Generics. FDA [Internet]. 2024 Feb 6 [cited 2024 Apr 23]; Available from: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/center-research-complex-generics
18. Research C for DE and. GDUFA II Pre-ANDA Program. FDA [Internet]. 2022 Sep 30 [cited 2024 Apr 23]; Available from: https://www.fda.gov/industry/generic-drug-user-fee-amendments/gdufa-ii-pre-anda-program
19. Research C for DE and. GDUFA III Enhancements to the Pre-ANDA Program. FDA [Internet]. 2023 Oct 10 [cited 2024 Apr 23]; Available from: https://www.fda.gov/industry/generic-drug-user-fee-amendments/gdufa-iii-enhancements-pre-anda-program
20. Yoon M, Babiskin A, Hu M, Wu F, Raney SG, Fang L, et al. Increasing impact of quantitative methods and modeling in establishment of bioequivalence and characterization of drug delivery. CPT Pharmacomet Syst Pharmacol. 2023 May;12(5):552–5.
21. Research C for DE and. Approved first generic for Apokyn injection cartridges requires separately packaged pen. FDA [Internet]. 2022 Feb 24 [cited 2024 Jun 26]; Available from: https://www.fda.gov/drugs/drug-safety-and-availability/approved-first-generic-apokyn-injection-cartridges-requires-separately-packaged-pen
22. Research C for DE and. 2022 First Generic Drug Approvals. FDA [Internet]. 2023 Mar 3 [cited 2024 Jun 26]; Available from: https://www.fda.gov/drugs/drug-and-biologic-approval-and-ind-activity-reports/2022-first-generic-drug-approvals
23. Research C for DE and. 2023 First generic drug approvals. FDA [Internet]. 2024 Mar 8 [cited 2024 Jun 26]; Available from: https://www.fda.gov/drugs/drug-and-biologic-approval-and-ind-activity-reports/2023-first-generic-drug-approvals
24. Addressing Barriers to the Development of Complex Generics: Understanding Challenges and Opportunities (White paper) .Complex Generics [Internet]. [cited 2024 Apr 29]. Available from: https://www.usp.org/complex-generics
25. Challenges in Complex Generic Drug Development [Internet]. DDReg Pharma. 2023 [cited 2024 Apr 29]. Available from: https://resource.ddregpharma.com/blogs/challenges-in-complex-generic-drug-development/
26. Gottlieb S. Reducing the Hurdles for Complex Generic Drug Development. FDA [Internet]. 2022 Mar 10 [cited 2024 Apr 29]; Available from: https://www.fda.gov/news-events/fda-voices/reducing-hurdles-complex-generic-drug-development
27. Stern S, Coghlan J, Krishnan V, Raney SG, Babiskin A, Jiang W, et al. Research and education needs for complex generics. Pharm Res. 2021 Dec;38(12):1991–2001.
28. Howard JN, Harris I, Frank G, Kiptanui Z, Qian J, Hansen R. Influencers of generic drug utilization: a systematic review. Res Social Adm Pharm. 2018 Jul;14(7):619–27.
29. Non-Biological Complex Drugs (NBCD) Working Group | Lygature [Internet]. 2021 [cited 2024 Apr 29]. Available from: https://www.lygature.org/non-biological-complex-drugs-nbcd-working-group
30. Expert PR. Complex Generics—Exploring the Challenges of Developing Complex Generics [Internet]. PDG. 2017 [cited 2023 Oct 17]. Available from: https://pharmdevgroup.com/breaking-down-complex-generics/
31. Rocco P, Musazzi UM, Franzè S, Minghetti P. Copies of nonbiological complex drugs: generic, hybrid or biosimilar? Drug Discov Today. 2019 Jan;24(1):250–5.
32. Musazzi UM, Marini V, Casiraghi A, Minghetti P. Is the European regulatory framework sufficient to assure the safety of citizens using health products containing nanomaterials? Drug Discov Today. 2017 Jun;22(6):870–82.
33. Gaspar RS, Silva-Lima B, Magro F,Alcobia A, da Costa FL and Feio J. Non-biological Complex Drugs(NBCDs): complex pharmaceuticals in need of individual robust clinical assessment before any therapeutic equivalence decision. Front Med. 2020;7:590527.
34. Katherine Tyner, PhD. An Overview of Complex Drug Substances and Complex Formulations—A Quality Perspective. Presentation to the Product Quality Research Institute. Available from: https://pqri. org/wp-content/uploads/2019/04/2-Tyner_PQRI.pdf
35. Guideline o the Investigation of Bioequivalence, accessed from EMA official website, Available from: http://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
36. Minghetti P, Musazzi UM, Casiraghi A, Rocco P. Old active ingredients in new medicinal products: is the regulatory path coherent with patients’ expectations? Drug Discov Today. 2020 Aug;25(8):1337–47.
37. Research C for DE and. The “Deemed to be a License” Provision of the BPCI Act: Questions and Answers [Internet]. FDA; 2020 [cited 2023 Dec 21]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deemed-be-license-provision-bpci-act-questions-and-answers
38. Guideline on similar biological medicinal products; EMA Science Medicines Health.2014 Oct 23;CHMP/437/04 Rev1. [Cited 2023 Dec 21]. Available from : https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
39. Lee S, Raw A, Yu L, Lionberger R, Ya N, Verthelyi D, et al. Scientific considerations in the review and approval of generic enoxaparin in the United States. Nat Biotechnol. 2013 Mar;31(3):220–6.
40. Falconer R, Jackson-Matthews D, Mahler S. Analytical strategies for assessing comparability of biosimilars. J Chem Technol Biotechnol. 2011 Jul 1;86:915–22.
41. Kozak D, Cai B, Babiskin A. Complex generic drug products (CGDPs) with complex formulations including nanotechnology products. Presentation at Association for Accessible Medicines (AAM): Generic + Biosimilar Medicines Conference (GBMC). Rockville, MD: Bethesda; 2019.
42. Emily M, Ioanna N, Scott B, Beat F. Reflections on FDA draft guidance for products containing nanomaterials: is the abbreviated new drug application (ANDA) a suitable pathway for nanomedicines? AAPS J. 2018 Sep;20(5):92.
43. Li D. Assessing API “Sameness”;Presentation at FDA CDER Small Business and Industry Assistance (SBIA):Advancing Generic Drug Development: translating science to approval Workshop; 21 Sep 2022.
44. Zhang D. Demonstrating Complex API Sameness; Presentation at FDA/CDER/OGD ; 06 Oct 2017.
45. Jiang X. Introduction to Complex Products and FDA Considerations. FDA[Internet].2017 Oct17 [cited on 17 Oct 2023]. Available from: https://www.fda.gov/files/drugs/published/Introduction-to-complex-products-and-FDA-considerations-Presentation.
46. Qu H, Wang J, Wu Y, Zheng J, Krishnaiah YSR, Absar M, et al. Asymmetric flow field flow fractionation for the characterization of globule size distribution in complex formulations: a cyclosporine ophthalmic emulsion case. Int J Pharm. 2018 Mar 1;538(1-2):215–22.
47. Burgess D. In vitro drug release from complex parenterals and development of IVIVCs. The Center for Research on Complex Generics (CRCG). [cited 2024 Mar 13]. Available from: https://complexgenerics.org/resource/in-vitro-drug-release-from-complex-parenterals-and-development-of-ivivcs/
48. Luke MC. Generic drugs for dermatology. The Center for Research on Complex Generics (CRCG). [cited 2024 Mar 13]. Available from: https://complexgenerics.org/resource/generic-drugs-for-dermatology/
49. Cilurzo F, Musazzi UM, Franzé S, Fedele G, Minghetti P. Design of in vitro skin permeation studies according to the EMA guideline on quality of transdermal patches. Eur J Pharm Sci. 2018 Dec 1;125:86–92.
50. Committee for Medicinal Products for Human Use. Guideline on Quality of Transdermal Patches. London, UK: Committee for Medicinal Products for Human Use; 2014.
51. Barton AE. Relevant challenges with IVRT with iron-carbohydrate complexes: Application to IVIVC models. The Center for Research on Complex Generics (CRCG). [cited 2024 Mar 13]. Available from:https://complexgenerics.org/resource/relevant-challenges-with-ivrt-with-iron-carbohydrate-complexes-application-to-ivivc-models/
52. O’Brien MN, Jiang W, Wang Y, Loffredo DM. Challenges and opportunities in the development of complex generic long-acting injectable drug products. J Controlled Release. 2021 Aug;336:144–58.
53. Bhattycharyya L, Dabbah R, Hauck W, Sheinin E, Yeoman L, Williams R. Equivalence studies for complex active ingredients and dosage forms. AAPS J. 2005 Dec;7(4):E786–812.
54. Sanduria S, Tripathy S, Dureja H. Voicing regulatory perspectives of the combination products. J Generic Med. 2020;16(3);101–11.
55. Lee SL, Saluja B, García-Arieta A, Santos GM, Li Y, Lu S, et al. Regulatory considerations for approval of generic inhalation drug products in the US, EU, Brazil, China, and India. AAPS J. 2015 Sep;17(5):1285–304.
56. Donnelly M, Tsakalozou E, Sharan S, Straubinger T, Bies R, Zhao L. Review of complex generic drugs delivered through the female reproductive tract: the current competitive landscape and emerging role of physiologically based pharmacokinetic modeling to support development and regulatory decisions. J Clin Pharm. 2020;60:S26–S33.
57. Kim MJ, Jarugula V. Clinical Pharmacology in Women’s Health: current Status and Opportunities. J Clin Pharmacol. 2020 Dec;60 Suppl 2:S7–S10.
58. Kapoor V, Kaushik D. A comparative study of regulatory prospects for drug-device combination products in major pharmaceutical jurisdictions. J Generic Med Sect. 2013 Jun;10(2):86–96.
59. Witzmann K. Complex generic drug-device inhalation products and user interface sameness: successful outcomes. The Center for Research on Complex Generics (CRCG). [cited 2024 Mar 13]. Available from: https://complexgenerics.org/resource/complex-generic-drug-device-inhalation-products-and-user-interface-sameness-successful-outcomes/
60. Kuribayashi R, Myoenzono A. First approval of generic dry powder inhaler drug products in Japan. Drug Deliv Transl Res. 2020 Oct;10(5):1517–9.
61. Regulatory Consideration for Approval of Generic Inhalation Drug Produ [Internet]. [cited 2023 Dec 21]. Available from: https://www.taylorfrancis.com/chapters/edit/10.1201/9781003046547-42/regulatory-consideration-approval-generic-inhalation-drug-products-us-eu-brazil-china-india-urvashi-parmar-jayvadan-patel
62. Research C for DE and. Office of generic drugs 2022 Annual Report. FDA [Internet]. 2023 Mar 1 [cited 2023 Oct 13]; Available from: https://www.fda.gov/drugs/generic-drugs/office-generic-drugs-2022-annual-report
63. Avhad PA, Chalikwar SS, Bhairav BA. Acomprehensive review on complex generics. Med Researchgate. 2022;105:129–35.
64. Complex generics: charting a new path [Internet]. [cited 2023 Jul 11]. Available from: https://www.iqvia.com/library/white-papers/complex-generics-charting-a-new-path
65. Zhang L, Lionberger RA. Generics 2030: where are we heading in 2030 for generic drug science, research, and regulation? Clin Pharmacol Ther. 2020 Jun;107(6):1293–5.
66. Pillsbury D. Certara. Advancing Biopharmaceutics and Drug Formulation Using In Silico Modeling. 2023 [cited 2023 Dec 21]. Available from: https://www.certara.com/white-paper/advancing-biopharmaceutics-drug-formulation-using-in-silico-modeling/
67. Troiano G, Nolan J, Parsons D, Van Geen Hoven C, Zale S. A quality by design approach to developing and manufacturing polymeric nanoparticle drug products. AAPS J. 2016 Nov;18(6):1354–65.
68. Lee J, Gong Y, Bhoopathy S, DiLiberti CE, Hooker AC, Rostami-Hodjegan A, et al. Public workshop summary report on fiscal year 2021 generic drug regulatory science initiatives: data analysis and model-based bioequivalence. Clin Pharmacol Ther. 2021 Nov;110(5):1190–5.
69. Lunawat S, Bhat K. Complex generic products: insight of current regulatory frameworks in US, EU and Canada and the need of harmonisation. Ther Innov Regul Sci. 2020 Sep;54(5):991–1000.
70. Bhatt M, Tank S, Shah J, Maheshwari D. Regulatory framework and disparities of complex generics in United States, European Union & Latin America. J Generic Med. 2023;19(3):130–40.
71. Kasuga M, Kuribayashi R, Ogawa T, Ugi A, Yamaguchi T, Takagi K, et al. Generic drug product development in Japan: regulatory updates during 2014–2019 and the future. Eur J Drug Metab Pharmacokinet. 2021 Nov;46(6):711–9.
72. Canada H. Policy Statement: Clarifying the appropriate regulatory pathway for subsequent entry low molecular weight heparins [Internet]. 2013 [cited 2023 Oct 13]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/policy-statement-clarifying-appropriate-regulatory-molecular-weight-heparins.html.
73. Pasam N, Kamaraj R. Regulation & registration of drugs and biologics in Brazil. J Med Pharm All Sci. 2022;11:4332–9.
74. Center for Drug Evaluation and Research (CDER). Determining whether to submit an ANDA or a 505(b)(2) application guidance for industry. [cited 2021 Jan 18]. Available from: https://www.fda.gov/media/124848/download
75. Government of India, Ministry of Health and Family Welfare. The drugs and cosmetics act, 1940 (as amended up to the 30th June, 2005) and The Drugs and Cosmetics Rules, 1945 (as amended up to the 30th June, 2005). [cited 2021 Jan 18]. Available from: http://cdsco.nic.in/ writereaddata/Drugs&CosmeticAct.pdf