
INTRODUCTION
Anogeissus dhofarica, a member of the Combretaceae family, is also known by the name Gahtti. It is a medium-sized deciduous tree that is 30 to 40 feet tall [1]. The genus Anogeissus contains eight species, five of which are native to South Asia, two of which are endemic to the southern Arabian Peninsula, and one of which is native to Africa [2].
Both Anogeissus bentii and A. dhofarica are unique to the southern Arabian Peninsula. The A. dhofarica tree is grown in Wadi Nahiz, Salalah, which is about a 12-km drive from Salalah city-Dhofar and situated on the north side of the city. Local Dhofarians have used the tree for thousands of years to treat sores and as a dye to color the coarse, unbleached cotton cloth. In addition, it was applied to wounds and used as an antiseptic, particularly by women for personal hygiene [3,4]. but the aforementioned claims are not backed up by any conclusive scientific data. Few pharmacological studies have been done on this plant, despite the fact that it is one of the most significant trees in Dhofar and has been used in a variety of ways [5], in contrast to other species such as Anogeissus latifolia, Anogeissus leiocarpous, and Alnus acuminate that have been thoroughly investigated [6-10].
Anogeissus dhofarica leaf extract underwent a high-resolution liquid chromatography/ultraviolet (HPLC/UV)-guided chemical analysis as part of our ongoing research on some medicinal plants of particular interest to Dhofarians [11-15].
Natural-derived materials, such as flavonoids, alkaloids, and polyphenols, have demonstrated significant anti-inflammatory activity, as well as low toxicity and increased pharmacological activity. As a result, these plant extracts are thought to be promising sources of natural anti-inflammatory medications. Recently, research into the role of these products in the treatment of inflammatory diseases has advanced significantly [16]. Prior research has focused on these products’ potential as anti-inflammatory treatments. Flavonoids were discovered to have a significant potential to stop cartilage deterioration, implying that they could be used to treat osteoarthritis [17]. Alkaloids are also known for their biological properties, such as anti-inflammatory properties. Previous research has shown that these compounds can slow the progression of rheumatoid arthritis (RA) in animal models [18]. Polyphenols have been shown to be anti-inflammatory in the treatment of inflammation-induced chronic atherosclerosis [18].
Therefore, we sought to evaluate how human dermal fibroblast proliferation and wound healing activity were impacted by A. dhofarica plant extracts. We also aimed to understand the role of inflammatory cytokines in the inflammatory response when cocultured with the human monocytic cell line THP-1 by observing the production of inflammatory cytokines.
MATERIALS AND METHODS
Plant material
Between July and September 2019, A. dhofarica leaves were collected in Wadi Nahiz valley, near Qairoon Hairiti on the northern side of Salalah city-Dhofar, about 19.00 km away (17.1895° N, 54.0861° E), Oman. In our laboratory, the leaves were shadow-dried at room temperature, macerated into a fine powder, and stored at room temperature. The A. dhofarica tree was classified by Alan Radcliffe-Smith (Royal Botanic Gardens, Kew, England). An A. dhofarica voucher specimen (VUA4 abcd) was deposited in the biology herbarium of Sultan Qaboos University in Muscat, Oman.
Preparation of A. dhofarica extracts
Ethanol, methanol, acetone, and ethyl acetate were among the polar and nonpolar solvents used. Each time, 500 ml of solvent was added to 100 g of finely ground leaf powder, and the mixture was stirred at room temperature for 12 to 14 hours. All extracts were collected, filtered through Whatman’s filter paper No. 1, and then concentrated under a vacuum using a rotary evaporator (Stuart, Cole-Parmer, NEO, Charlotte ST, UK). Based on their boiling points, the solvents evaporated at temperatures ranging from 60°C to 80°C. Evaporation temperatures for acetone and methanol ranged from 60°C to 67°C, whereas ethanol and ethyl acetate ranged from 78°C to 80°C.
The powdered materials were stored in covered containers at room temperature. The extracts of A1 (100% ethanol), A3 (100% methanol), A5 (acetone), and A8 (ethyl acetate) were coded for the powdered samples. Methanol and ethanol extracts (61 and 55 mg/g, respectively) recovered the most solid material. The least amount of solid material was produced by ethyl acetate and acetone extracts (46 and 39.5 mg/gm, respectively).
Extract preparation
Plant extracts (A1, A3, A5, and A8) were dissolved in 1 ml of DMSO to achieve a concentration of 100 mg/ml. Ethanol extracts (A1) were dissolved in 1 ml of cell culture medium (DMEM, Gibco, Carlsbad, CA) at this time. Following the preparation of 30 mg/ml from 100 mg/ml, a series of dilutions were carried out to produce extracts at different concentrations of 3, 0.3, and 0.03 mg/ml.
Total phenolic content
The Folin–Ciocalteu method was used to calculate the total phenolic content. In this procedure, a test tube containing 750 µl of Na2CO3 (1.9 M) and 250 µl of Folin–Ciocalteu reagent was added with 15 µl of blank, standard, or diluted extract. The total volume was 5 ml of distilled water. The tubes were continuously vortexed for 1 min before being incubated at room temperature without light for 2 hours. After that, an absorbance measurement at 765 nm was performed using a spectrophotometer (He?IOS α). The calibration curve was generated using typical gallic acid solutions. Gallic acid equivalents in mg/g of sample were used to express the results [16].
Total flavonoid content
Five hundred microliters of each standard or diluted sample was added to a test tube containing 2 ml of distilled water, 0.15 ml of sodium nitrite, and 0.15 ml of 10% aluminum chloride solution (Sigma-Aldrich, St. Louis, MO). Each sample was supplied with 1 ml of 1 M sodium hydroxide (Sigma-Aldrich, St. Louis, MO) after 5 minutes of incubation. The reaction tube was diluted with the addition of 1.2 ml of distilled water (to make the total volume up to 5 ml) and thoroughly mixed for 30 seconds. Finally, a spectrophotometer (He?IOS α) was used to measure the pink mixture’s absorbance at 510 nm. Different concentrations of the catechin standard solution were used to construct the calibration curve, and the results were expressed as catechin equivalents in mg/g of sample [13].
Determination of active phytochemicals by using high-performance liquid chromatography (HPLC-DAD)
Seven phenolic standard compounds (ferulic acid, caffeic acid, gallic acid, o-coumaric acid, m-coumaric acid, 3,4-dihydroxybenzoic acid, and syringic acid) were prepared and used for quantitative analysis with HPLC-DAD.
Stock standard solutions of each compound were prepared in ethanol or water at a concentration of 100 mg/l (100 ppm) for the linearity study. All samples (0.02 g diluted in 1 ml of solvent) and standards were filtered into standard HPLC vials using a 0.45 µl membrane filter before HPLC analysis. The volume of the injected sample was 10 μl and the mobile phase contained methanol:water:formic acid (10:88:2, v/v/v) (solvent A) and methanol:water:formic acid (90:8:2, v/v/v) (solvent B) with a flow rate of 0.5 μl/minutes. Then, a 300 × 3.9 mm, 15–20 µm ODS column (HiChrom, Lutterworth, UK) was used in conjunction with a DAD product to carry out the chromatographic separation.
Fibroblast viability
A total of 5 × 103 fibroblasts (ATCC, Manassas, VA) were planted into a 96-well plate (SPL, Gyeonggi-do, Korea) and allowed to adhere for 24 hours before being treated with various concentrations of A. dhofarica plant extract: 30, 3, 0.3, 0.03 mg/ml, and a blank control (DMEM with 10% fetal bovine serum (FBS) only (Gibco, Carlsbad, CA), then incubated at 37°C for 72 hours in a 5% CO2 incubator. After incubation, the old medium was substituted with 100 µl of fresh medium and 15 µl of MTT [3-[4,5-dimethyl-2thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide] (5 mg/ml) (Promega, Madison, WI) was added to all wells, and the plates were then left to incubate at 37°C for 3 hours. 50 µl of DMSO was added to each well to halt the process. An optical density (O.D.) was measured at 570 nm after 10 minutes using a Glomax plate reader (Promega, Madison, WI). Nonlinear regression and GraphPad Prism were used to calculate the IC50.
Apoptosis/necrosis cell death modality
To see if our treatments had any effect on the mode of cell death, we used an apoptosis/necrosis assay. First, 1 × 105 fibroblasts were planted into a 6-well tissue culture plate (SPL, Gyeonggi-do, Korea). The cells were subsequently incubated for 72 hours with A. dhofarica plant extracts at the following concentrations: 30, 3, 0.3, and 0.03 mg/ml. Cells were isolated and collected using trypsin EDTA 1x (Biowest, France) after 72 hours of treatment. The cells were then centrifuged for 5 minutes at 300 × g after being cleaned with PBS (Gibco, Carlsbad, CA). An apoptosis kit (Invitrogen, Waltham, MA) with Annexin V/PI stain was then used to stain the cells. The samples were examined using FACS DIVA 7 software and FACS Canto11 (BD Biosciences, Franklin Lakes, NJ).
Inflammatory response-THP-1 cell activation
The human monocytic cell line THP-1 (ATCC, Manassas, VA) was maintained in RPMI 1640 cell culture growth medium (Euroclone, Milano, IT) containing the following: 10% FBS and 4.5 g/l D-glucose (Sigma. St. Louis, MO), 1X L-glutamine, and 1X penicillin/streptomycin (Biowest, Nuaillé, France). As previously described [17], THP-1 cells were treated for 24 hours with 100 nM phorbol 12-myristate 12-acetate (PMA; Biotechne, Minneapolis, NE) at a concentration of 2 × 105 cells per ml to differentiate them into macrophage. Nonadherent cells were aspirated from the plate after incubation, and differentiated adherent cells were cultured in a cell growth medium. Adherent macrophages were stimulated by lipopolysaccharide (5 g/ml LPS, Santa Cruz Biotechnology, Santa Cruz, CA) treatment for 24 hours. After the cells had been activated for 24 hours, they were treated for another 24 hours with RPMI supplemented with various plant extract concentrations (3, 0.3, and 0.03 mg/ml). The control cells, adherent macrophages, were only activated by lipopolysaccharide without receiving varying concentrations of plant extract.
Quantification of cytokines by the cytometric beads array (CBA) multiplex method
A panel of cytokines (IL-1β, TNF-α, IL-6, IL-8, IL-10, and IL-12p70) was detected and measured by flow cytometry using human inflammatory cytokine CBA array beads (BD Biosciences, Franklin Lakes, NJ) to determine the impact of extracts on macrophages. In brief, media were collected and stored at −80°C until use after 24 hours of treatment with different concentrations of plant extracts (3, 0.3, and 0.03 mg/ml). Cells that had not been exposed to plant extracts served as the negative controls. The samples were then prepared and examined in accordance with the manufacturer’s instructions. The analysis was conducted using a FACS Canto II (BD Biosciences, Franklin Lakes, NJ).
Wound healing assay
Scratch assay
The scratch assay was performed as previously described [18]. Briefly, fibroblastic cells were seeded into 6-well culture plates (SPL, Gyeonggi-do, Korea) and allowed to reach 100% confluence. Confluent cells were then incubated in serum-free medium for 24 hours. to starve them. After 24 hours, 200 µl pipettes (Diamond Tipack, 20–200 µl Gilson, Middleton, WI) were used to make a vertical scratch on the confluent layer of cells in each well. After the serum-free medium was removed, the cells were washed with PBS to remove any remaining cell debris. They were then incubated for 72 hours. in DMEM with various concentrations of extracts (A1, A3, A5, and A8) at 3, 0.3, and 0.03 mg/ml as well as a blank control.
Microphotographs of the scratch were taken at two different time intervals: 0 and 72 hours. The cultured cells were then examined under an inverted microscope (Axiovert, Zeiss, Oberkochen, Germany) to look for variations in their closure pattern.
Transwell migration assay
The second technique for assessing cell migration involved the use of two chamber Transwell systems (Costar, Corning, NY) with inserts that were 6.5 mm in diameter and 8 µm pore size.
In the upper chamber, 5 × 104 cells were resuspended in 250 µl DMEM, and 700 µl of media-containing various treatment concentrations were added to the lower chamber. After 24 hours, the migrated cells that crossed the membrane to the bottom chamber were fixed for 10 minutes, in 4% paraformaldehyde (Sigma, St. Louis, MO). After being incubated with the molecular probe DAPI (Gibco, Carlsbad, CA) for five minutes, the membrane was taken out and mounted to a drop of mounting medium (DAKO, Troy, MI) using glass slabs before being covered with yet another cover slip. To quantify the migrated cells, a fluorescence microscope (Axio Imager z2, Zeiss, Oberkochen, Germany) was used to count 10 randomly selected microscopic fields per filter at 200× magnification. Measurements were made in triplicate for each experiment, and the results were calculated as the mean.
Statistical Analysis
To determine the significant differences among all assays, the results were analyzed using GraphPad Prism 6 (San Diego, CA) and Microsoft Windows Excel. In three dependent experiments, all experiments were performed in triplicate (n = 3). The IC50 was determined using nonlinear regression analysis for all plant extracts. Two-way ANOVA and Tukey’s post-test were used to analyze the results of the transwell migration assays, the inflammatory response, and the cell death analysis. The data were considered significant if p <0.00005***, p = 0.0005***, p < 0.005**, and p < 0.05*. All data are displayed as the mean ± SD.
RESULTS AND DISCUSSION
Recent studies have concentrated on using natural products as alternatives to synthetic therapeutic drugs. Natural products have a vast array of potential applications in the pharmaceutical industry because of their accessibility, diversity, and chemical composition [19]. In addition, there is a critical need to find new natural products with high therapeutic potential because existing chemotherapeutic drugs and antibiotics are hindered by adverse events and/or the loss of efficacy, particularly in the latter case due to the development of pathogen resistance. To find new sources of medicinal agents, there is a high demand for screening various phytochemical compounds extracted from various medicinal plants with unknown pharmacological activities [20,21]. The present study aimed to investigate the phytochemical, biological, and potential therapeutic applications of leaf extracts from A. dhofarica.
Determination of active phytochemical compounds by using high-performance liquid chromatography (HPLC)
Using an HPLC method, the total phenolic compounds found in this study were analyzed. Seven phenolic compounds (ferulic acid, caffeic acid, gallic acid, o-coumaric acid, m-coumaric acid, 3,4-dihydroxybenzoic acid, and syringic acid) were utilized for the quantitative analysis of phenolics. Retention time and the absorbance spectrum profile were used to identify phenolic compounds in relation to a standard comparison of each identified compound. The peak areas of extracted samples were used to calculate the phenolic chemicals’ retention times (Table 1). Olive oil, carrots, tomatoes, coffee, and fruit juices are just a few examples of foods and beverages that naturally contain the water-soluble phenolic compound known as caffeine. It has properties that are anti-oxidant, anti-mutagenic, anti-tumor, and anti-obesity [22].
 | Table 1. Retention time (RTS), absorbance at ? 210 nm based on the area under the curve. [Click here to view] |
A. dhofarica extracts in ethanol, methanol, acetone, and ethyl acetate were recently subjected to a preliminary phytochemical screening using an HPLC/UV-guided method (Fig. 1). The results showed that all four extracts contain significant levels of total phenolic compounds, ranging from 188.5 to 544.3 mg/g. Total flavonoid compounds ranged from 96.1 to 398.9 mg/g (Table 2). Total phenolic compound concentrations were highest in the ethanolic extract and lowest in the acetone extract (Table 2). With the exception of the ethyl acetate extract, which has the highest flavonoid content (398.9 mg/g), all extracts typically contain fewer flavonoids than phenolic compounds (Table 2).
The phenolic concentration varied significantly among the four extraction solvents. For instance, the highest phenolic concentrations were found in the ethyl acetate and acetone extracts, at 119.195 and 43.047 mg/ml, respectively (Table 3). The lowest phenolic concentrations, 3.601 and 0.615 mg/ml, were found in the methanolic and ethanolic extracts, respectively (Table 3). Finally, the present study demonstrated that the major components present in the leaves of A. dhofarica are phenolic compounds and flavonoids. These findings are consistent with those of [23] and [24] on A. leiocarpus.
The isolated phenolic compounds and flavonoids from the genus Anogeissus both have biological properties that are very intriguing. They exhibit antibacterial, antiviral, immune, anti-inflammatory, wound-healing, antioxidant, cytotoxic, and antitumor activity via the angiogenesis pathway, and they have been shown to interfere with a variety of physiological events both in vitro and in vivo [25–28].
Cell viability assay (MTT)
An MTT assay was used to calculate the IC50 value of the plant extracts on fibroblasts after 72 hours. Plant extracts of varying concentrations were applied to fibroblasts under the same conditions as the untreated control cells. The calculated IC50 values for plant extracts A1, A3, A5, and A8 are 9.799 ± 0.07, 5.83 ± 0.9, 8.8 ± 0.12, and 3.98 ± 0.8 mg/ml, respectively, as shown in Fig. 2.
Apoptosis/necrosis cell death modality
To ascertain whether our treatments had any impact on the mode of cell death, an apoptosis/necrosis assay was used. According to our data, ethanolic extract (A1) significantly increased the number of apoptotic/necrotic cells compared to the untreated control groups (p < 0.0005). The percentages of apoptotic/necrotic cells for the methanolic extract (A3) were significantly different in all treated groups (3, 0.3, and 0.3 mg/ml) compared to the untreated group (p < 0.0005).
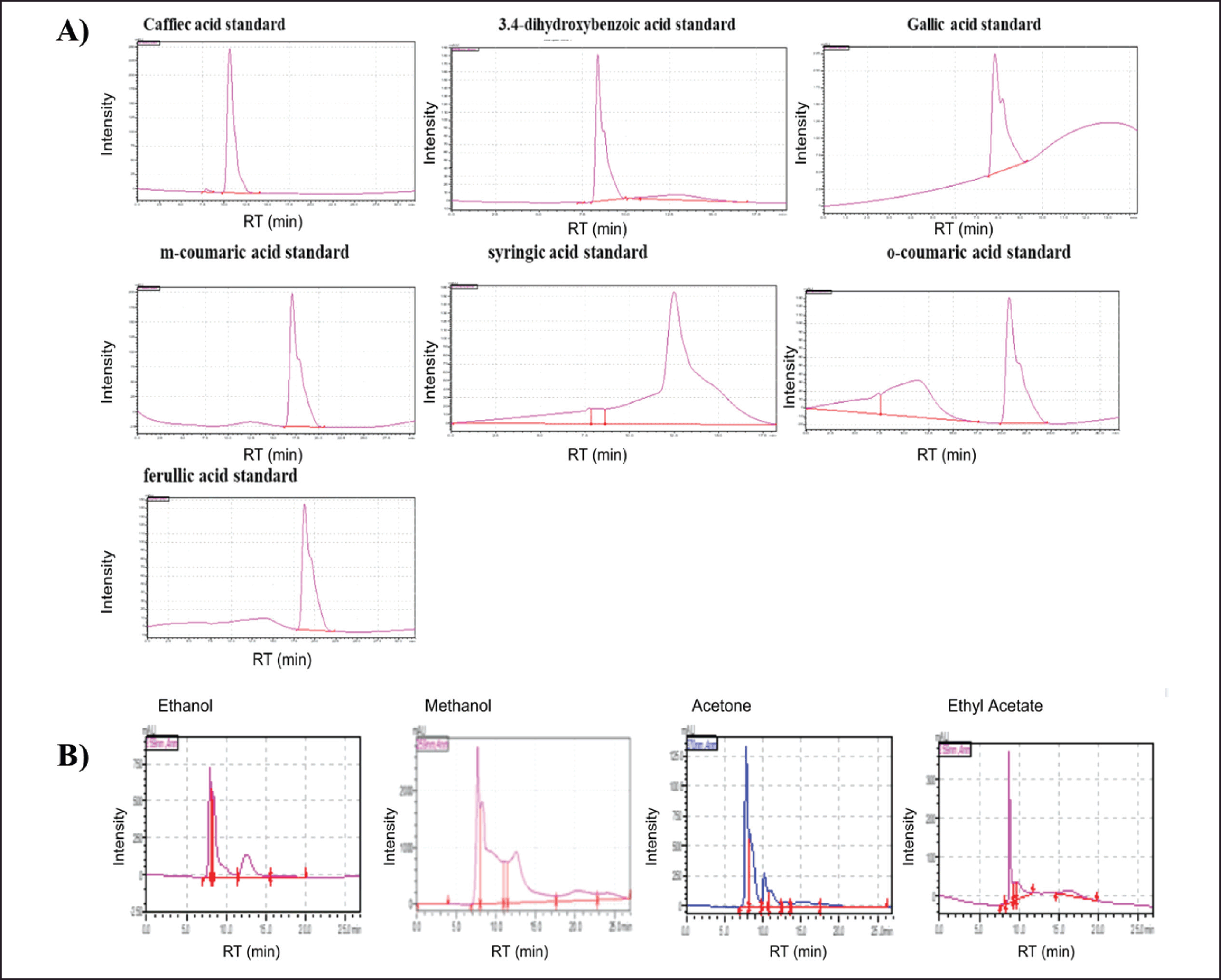 | Figure 1. HPLC-DAD-MS/MS chromatograms of Phenolic standard compounds (ferulic acid, caffeic acid, gallic acid, o-coumaric acid, m-coumaric acid, 3,4-dihydroxybenzoic acid, and syringic acid) in A. dhofarica extracts (A). A. dhofarica extracts in ethanol, methanol, acetone, and ethyl acetate showed that all four extracts contain significant levels of total phenolic compounds using an HPLC/UV-guided method (B). [Click here to view] |
Similarly, after treating cells with 3 and 0.3 mg/ml of the A5 extracts, the percentages of apoptotic/necrotic cells changed significantly (p < 0.0005). In contrast to the control group, 0.03 mg/ml A5 did not exhibit any cytotoxic effects. However, all concentrations of the A8 ethyl acetate extract considerably raised the percentages of apoptotic/necrotic cells in comparison to the untreated group (p < 0.0005).
As a result, with the exception of the lowest concentration of the acetone extract, all extract concentrations were significantly higher in late apoptotic cells than in the untreated group (Fig. 3).
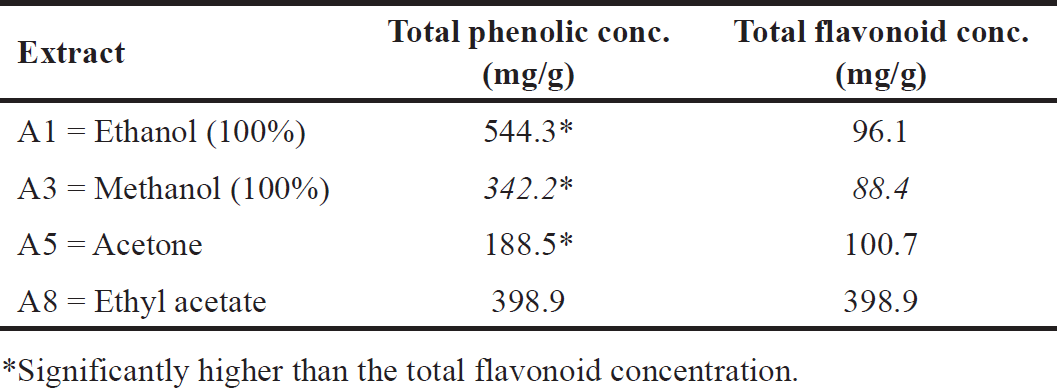 | Table 2. Total phenolic and flavonoid content of A. dhofarica extracts. [Click here to view] |
Based on the IC50 values for the cytotoxic and apoptotic effects of the four extracts, which ranged from 3.98 to 9.799 mg/ml, we discovered that the ethyl extract was the least cytotoxic and the ethanolic extract was the most cytotoxic. The results of this study are consistent with those of [26,29], who discovered that all extracts obtained from A. dhofarica were cytotoxic to brine shrimp larvae. However, our study focused on the cytotoxic effect on normal fibroblast cells, whereas previous studies used cancer cells to assess toxicity [26,29]. Furthermore, when compared to other extracts such as Desmodium adscendens, our A. dhofarica extracts were less toxic [30].
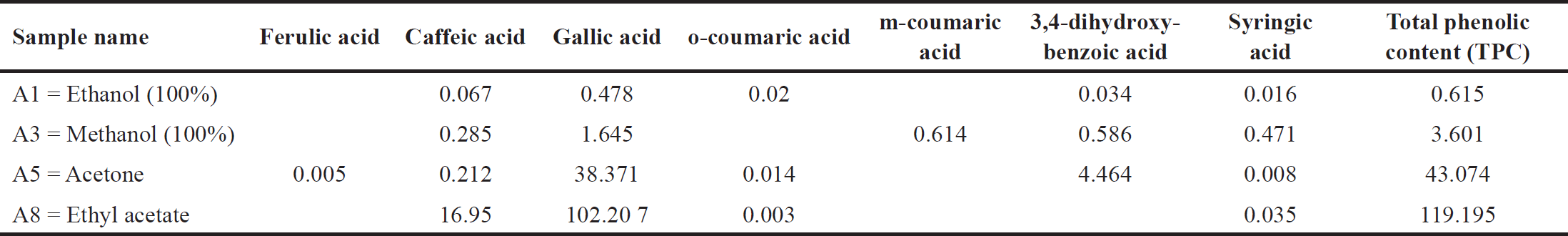 | Table 3. Phenolic concentration (mg/g) A. dhofarica extracts. [Click here to view] |
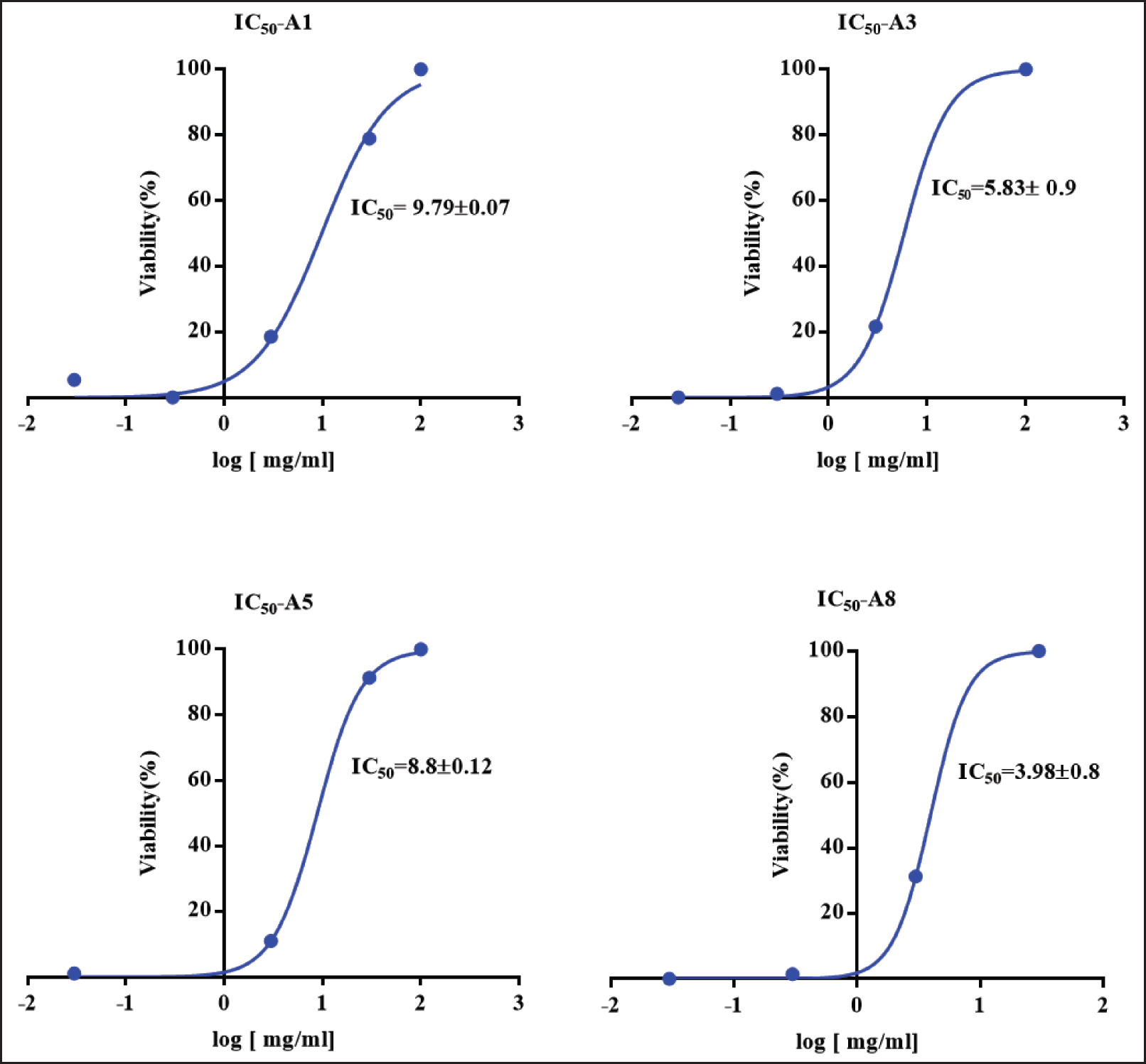 | Figure 2. IC50 values for the fibroblast viability after 72 hours of treatment with A. dhofarica extracts as determined by MTT assay. A1 (ethanol), A3 (methanol), A5 (acetone), and A8 (ethylacetate). Data were collected as triplicates from three independent experiments. [Click here to view] |
Anti-inflammatory response
The secretion of a panel of cytokines (IL-1, IL-6, IL-8, IL-10, IL-12p70, and TNF-α) after macrophage activation in the presence and absence of various concentrations of plant extracts was assessed by flow cytometry using CBA array beads. In comparison to the control group (macrophages treated with LPS), all plant extracts significantly increased the secretion levels of IL-1, IL-6, and TNF-α (p < 0.0005).
In addition, IL-8 was significantly upregulated (p <0.0005) among all concentrations, except 3 mg/ml of the acetone extract (A5), where the secretion level was significantly downregulated (p < 0.0005) compared to the control group. The maximum concentration (3 mg/ml) of all plant extracts was able to significantly upregulate (p < 0.0005) IL-12p70 secretion when compared to the control group. The doses of 0.3 and 0.03 mg/ml significantly boosted cytokine secretion to a lesser extent with methanol (A3) (p < 0.0005, p < 0.005) and acetone extract (A5) (p < 0.05, p < 0.005), respectively. The expression of IL-10, however, was significantly reduced by all treatments (p < 0.0005) (Table 4).
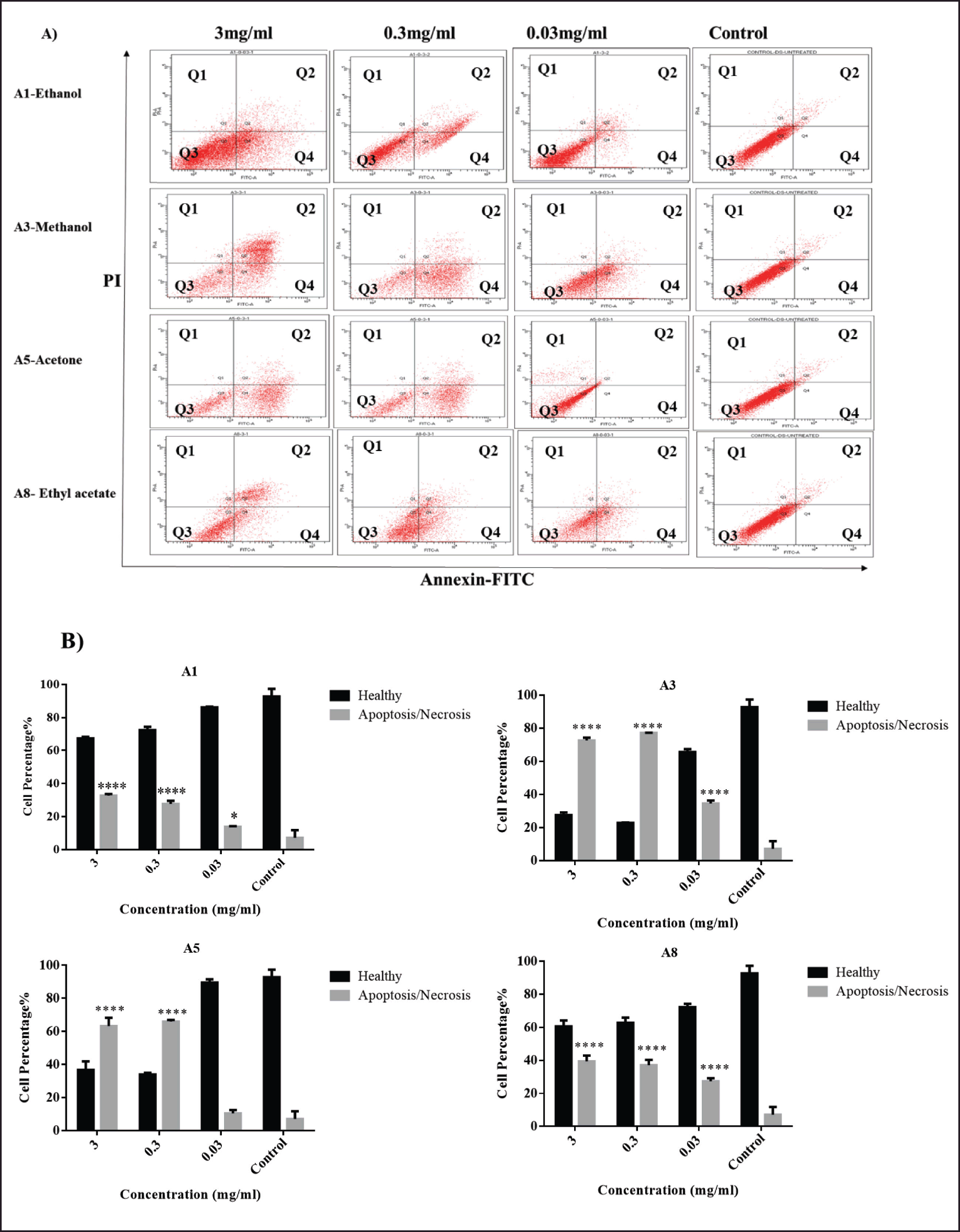 | Figure 3. Flow cytometric cell death analysis. (A) Flow cytometric dot plots and (B) Statistical analysis of fibroblast cells, treated with 3, 0.3, and 0.03 mg/ml of the following A. dhofarica extracts: A1 (ethanol), A3 (methanol), A5 (acetone), and A8 (ethylacetate) for 72 hours, and compared to the control untreated cells. Q1: necrosis, Q2: Late apoptosis, Q3: Healthy cells, and Q4: Early Apoptosis. Data were collected as triplicates from three independent experiments. ****p < 0.005, *p < 0.05. [Click here to view] |
Hence, based on the upregulation of the secretion of the aforementioned cytokines, our findings thus suggested that our plant extracts were liable for the differentiation of macrophages into the M1 subtype, in which M1 macrophages have bactericidal activity and are involved in the mechanism of tissue repair and regeneration [31,32]. These results are consistent with previous studies that have confirmed the antibacterial activity of these extracts [26,29,30]. Future research on both extracts A3 and A5 will be needed, but at concentrations other than those employed in this study.
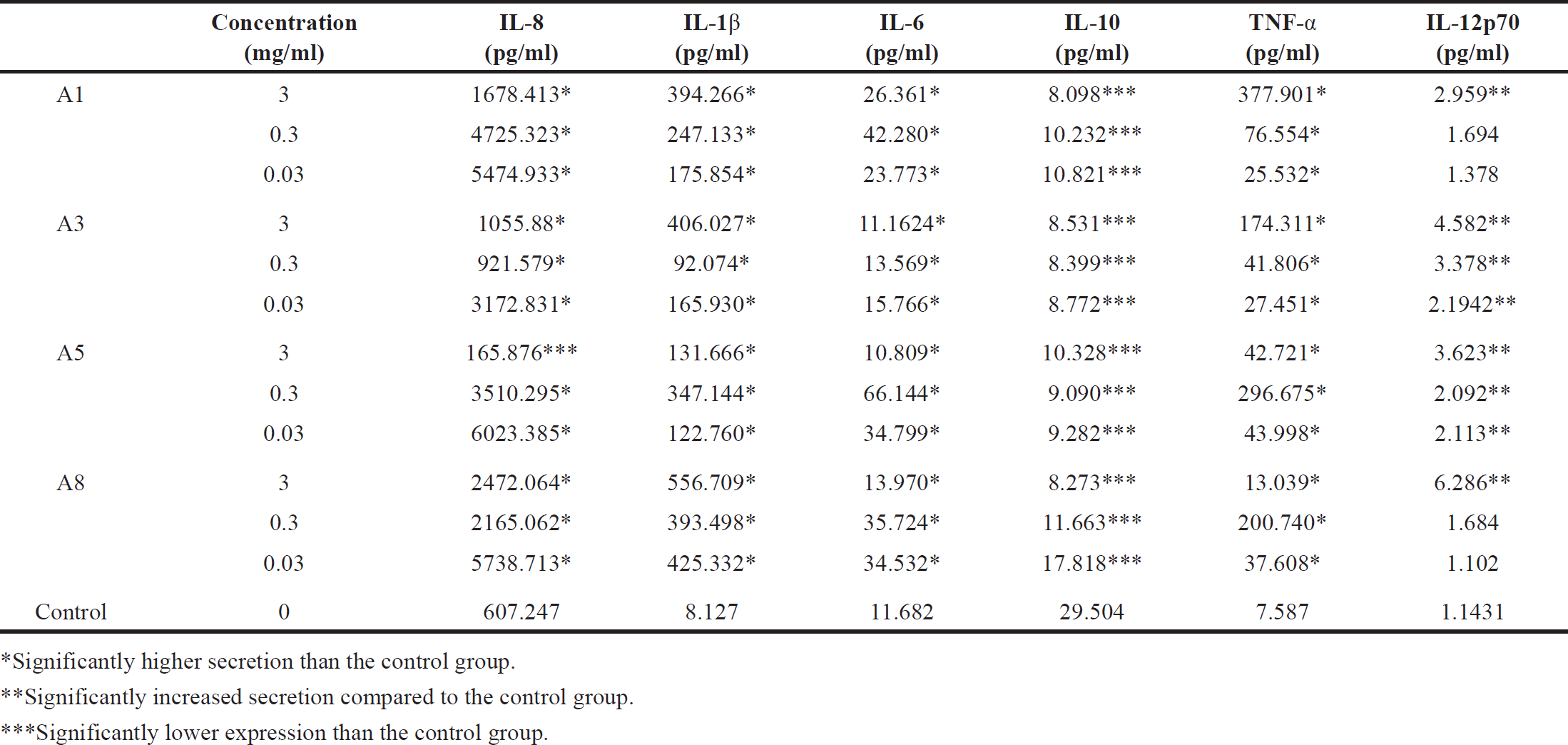 | Table 4. Measurements of the secretion levels of pro- and anti-inflammatory cytokines by multiplex bead array assays: (IL-1β, TNF-α, IL-6, IL-8, IL-10, and IL-12p70), following PMA activation of macrophages (differentiated THP-1 cells), treated with different concentrations; 3, 0.3, and 0.03 mg/ml of extracts. A. dhofarica extracts: A1 (ethanol), A3 (methanol), A5 (acetone), and A8 (ethyl acetate), compared to untreated control cells (cells that had not been treated with plant extracts). Data were collected as triplicates from three independent experiments. [Click here to view] |
 | Figure 4. Wound healing (scratch assay). Photomicrographs of fibroblast cells treated with different concentrations; 3, 0.3, and 0.03 mg/ml of A. dhofarica extracts: A1 (ethanol), A3 (methanol), A5 (acetone), and A8 (ethyl acetate) compared to the untreated control cells. Photomicrographs were taken at two different time points, 0 and 72 hrs after inflicting the wound. [Click here to view] |
Wound healing assay
Wound healing is a physiological response to tissue damage caused by an injury. It triggers tissue regeneration and wound contraction until the damaged tissue’s tensile strength is restored and repaired.
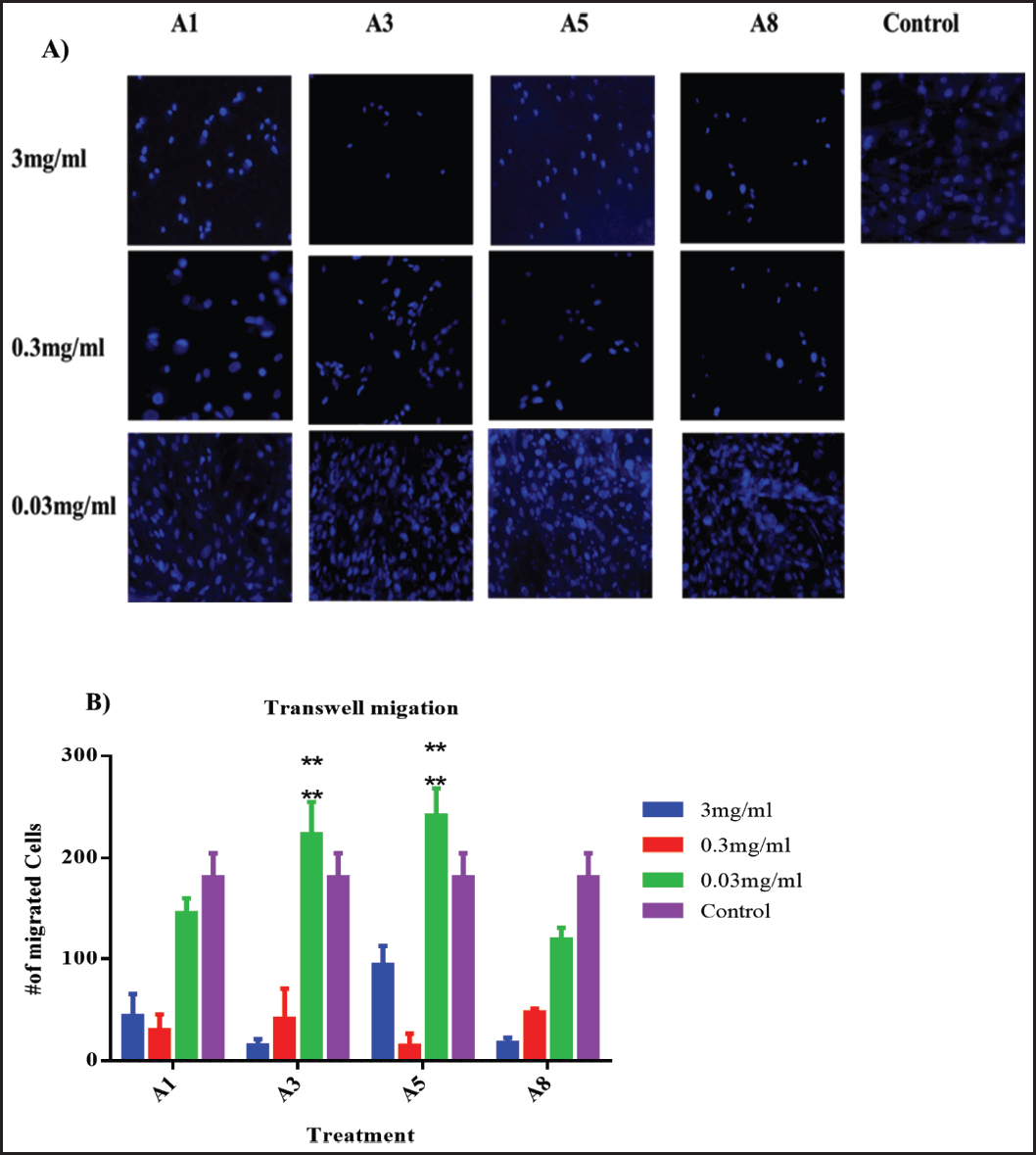 | Figure 5. Two-chamber Transwell system. (A) Photomicrographs of representative samples. (B) Statistical analysis of migrating fibroblast cells treated with different concentrations, i.e., 3, 0.3, and 0.03 mg/ml of A. dhofarica extracts: A1 (ethanol), A3 (methanol), A5 (Acetone), and A8 (Ethyl acetate), compared to the control untreated cells after 24 hours of treatment. Experiments were performed in triplicates and 10 random fields were counted on each membrane by using a fluorescence microscope. ****p < 0.005. [Click here to view] |
This outcome is the result of a complex chain of well-ordered biochemical and cellular reactions involving various tissues and cells. For example, during skin wound healing, there are phases of inflammation, proliferation, and maturation [26,29,31]. To assess the impact of our treatments on fibroblast wound healing, we conducted a Transwell migration experiment as well as a wound healing (scratch) assay.
After 72 hours of treatment with ethyl acetate plant extract (A8), the scratch wound was able to heal at concentrations of 0.3 and 0.03 mg/ml. Furthermore, 0.03 mg/ml of acetone (A5) and 0.03 mg/ml of methanol (A3) extracts completely healed the scratch. The ethanolic plant extract (A1), on the other hand, was unable to heal the scratch (Fig. 4). Furthermore, using the Transwell system (two chambers), we investigated the effect of our treatments on the potential for vertical migration within tissues.
Low concentrations (0.03 mg/ml) of both acetone (A5) and methanol (A3) extracts from plants showed a highly significant (p < 0.0005) potential for migration when compared to the untreated control group. All other plant extracts and concentrations, however, had fewer migrating cells compared to the control group (Fig. 5).
Given the effects of the four extracts on cytokines, which represent their anti-inflammatory potential, it currently seems difficult to determine which of the four extracts inhibits both wound healing and Transwell assays. The polarity of the solvents used in the four extracts, however, might have also been a factor. More polar and nonpolar solvent assays are required to further explore this activity because these appear to be the most contentious findings in the current study.
In summary, this is the first study to investigate the impact of A. dhofarica extracts on the biological processes of human normal fibroblast cells, specifically proliferation, cytotoxicity, wound healing, and inflammatory response. Previous studies have shown that these extracts have anticancer activity against a variety of cell lines, including MDA-MB. In addition to their antibacterial, antifungal, and antioxidant properties [26,28,30].
CONCLUSION
The methods we assessed for this study provide information about A. dhofarica extracts’ potential medicinal applications in wound healing, cytotoxicity, and inflammatory response, among other fields. Our findings concluded that lower concentrations of the plant extract had the least cytotoxic effect and the greatest wound healing activity, in addition to modulating inflammation in vitro by increasing the secretion levels of the following cytokines: IL-1, IL-6, TNF-, IL-8, and IL-12p70.
Future work
Despite this, the current study’s findings are intriguing and warrant further investigation, including the use of more polar and nonpolar solvents, as well as focusing on some of those extracts that can be described as biocompatible materials and purifying them to identify some pure compounds via bioactivity-guided fractionation. More research is needed to determine the therapeutic benefits of acetone and methanolic extracts on living organisms. In addition, further research employing molecular modeling experiments should be conducted to provide useful insights into the structure and function of molecules, allowing for a better understanding of the mechanisms underlying the extract’s biological action.
LIST OF ABBREVIATIONS
A. dhofarica: Anogeissus dhofarica; FBS: fetal bovine serum; MTT: [3-[4,5-dimethyl-2thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide]; CBA: Cytometric beads array; IL: interleukin; TNF-α: tumor necrosis factor-alpha.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
The authors would like to thank the Deanship of Scientific Research at the University of Petra for its support with Project No.: 36/4/2020.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests to disclose.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated or analyzed during this study are included in this published article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Kirtikar KR, Basu, BD, Basu LM. Indian medicinal plant part 1-3. Allahabad, India: Bishan Singh Mahendra Agency; 1935.
2. Kumaraswamy M, Kavitha H, Satish S, Antibacterial potential of extracts of Woodfordia fruticosa Kurz. on human pathogens. World J Med Sci. 2008;3(2):93–6.
3. Miller AG, Morris M. Plants of Dhofar (the southern region of Oman: traditional, economic and medicinal uses). The Office of the Advisor for Conservation of the Environment, Diwan of Royal Court, Sultanate of Oman. Holmes McDougall Ltd., Allander House, Edinburgh, UK; 1988. https://doi.org/10.1017/S0030605300035006
4. Singh D, Baghel US, Gautam A, Baghel DS, Yadav D, Malik J, et al. The genus Anogeissus: a review on ethnopharmacology, phytochemistry, and pharmacology. J Ethnopharmacol. 2016;194:30–56. CrossRef
5. Dubey D, Patnaik R, Ghosh G, Padhy RN. In vitro antibacterial activity, gas chromatography–mass spectrometry analysis of Woodfordia fruticosa Kurz. leaf extract and host toxicity testing with in vitro cultured lymphocytes from human umbilical cord blood. Osong Public Health Res Perspect. 2014;5(5):298–312.
6. Dabur R, Gupta A, Mandal T, Singh DD, Bajpai V, Gurav A, et al. Antimicrobial activity of some Indian medicinal plants. Afr J of Tradit Complement Altern Med. 2007;4(3):313–8.
7. Romulo A, Zuhud EA, Rondevaldova J, Kokoska L. Screening of in vitro antimicrobial activity of plants used in traditional Indonesian medicine. Pharm Biol. 2018;56(1):287–93.
8. Velmurugan B, Suresh M. Bioactive compound separation and antimicrobial activity of Ipomoea aquatica FORSK. Uttar Pradesh J Zool. 2020;41(21):40–8.
9. Parekh J, Chanda S. In vitro antibacterial activity of the crude methanol extract of Woodfordia fruticosa Kurz. flower (Lythraceae). Braz J Microbiol. 2007;38:204–7.
10. Indukuri R, Bhanu P, Vattipalli M. Bhargavi Ch. wound healing activity of leaf extract of Woodfordia fruticosa in albino rats. Int J Phytopharmacol. 2014;5(2):296–304..
11. Raghuwanshi N, Kumari P, Srivastava AK, Vashisth P, Yadav TC, Prasad R, et al. Synergistic effects of Woodfordia fruticosa gold nanoparticles in preventing microbial adhesion and accelerating wound healing in Wistar albino rats in vivo. Mater Sci Eng. C. 2017;80:252–62.
12. Ouedraogo V, Kiendrebeogo M. Methanol extract from Anogeissus leiocarpus (DC) Guill. et Perr.(Combretaceae) stem bark quenches the quorum sensing of Pseudomonas aeruginosa PAO1. Medicines. 2016;3(4):26.
13. Govindarajan R, Vijayakumar M, Singh M, Rao CV, Shirwaikar A, Rawat A, et al. Antiulcer and antimicrobial activity of Anogeissus latifolia. J Ethnopharmacol. 2006;106(1):57–61.
14. Elsiddig IME, Muddather AK, Ali HAR, Ayoub SMH. A comparative study of antimicrobial activity of the extracts from root, leaf and stem of Anogeissus leiocarpous growing in Sudan. J Pharmacogn Phytochem. 2015;4(4):107.
15. Warrier PK. Indian medicinal plants: a compendium of 500 species. Chennai, India: Orient Blackswan; 1993. Vol. 5.
16. Yadav S, Sardesai M. Flora of Kolhapur district. 2002.
17. Abuarqoub D, Mahmoud NN, Zaza R, Abu-Dahab R, Khalil EA, Sabbah DA. The in vitro immunomodulatory effects of gold nanocomplex on THP-1-derived macrophages. J Immunol Res. 2022;2022:1–7.
18. Abuarqoub D, Aslam N, Jafar H, Abu Harfil Z, Awidi A. Biocompatibility of biodentine™® with periodontal ligament stem cells: in vitro study. Dent J. 2020;8(1):17.
19. Rojas R, Bustamante B, Bauer J, Fernández I, Albán J, Lock O. Antimicrobial activity of selected Peruvian medicinal plants. J Ethnopharmacol. 2003;88(2–3):199–204.
20. Tyler VE. Chapter 3 Phytomedicines in Western Europe: potential impact on herbal medicine in the United States. 1993. pp. 25–37.
21. Al-Othman AM, Hussain I, Khan H, Rehman MU, Abdeltawab AA, Ullah R, et al. Phytochemical analysis and biological activities of selected medicinal plants. J Med Plants Res. 2012;6(23):4005–4010.
22. Collins HR. Caffeic acid: sources, potential uses and health benefits. New York, NY: Nova Science Publishers Incorporated; 2017.
23. Mann A, Amupitan J, Oyewale A, Okogun J, Ibrahim K. Antimicrobial potentials of Anogeissus leiocarpus and Terminalia avicennioides in treating infectious diseases prevalent in Hospital Environments in Nigeria. J Women Technol Educ Employment. 2007;5(1):67–74.
24. Adeleye I, Ogunniyi A, Omonigbehin E. Antimicrobial activity of some local herbs on common skin pathogens. Biosci Res J. 2021;15(3):22–7..
25. Hassana LEA, Al-Suadea F, Fadul SM, Majida A. Evaluation of antioxidant, antiangiogenic and antitumor properties of Anogeissus leiocarpus against colon cancer. Angiotherapy. 2018;1(2):56–66.
26. Al-Noumani AJ, Al-Qasmi MZ, Al-Shabibi AS, Al-Mashani SA, Alabri AA, Sohailakhtar M. Antimicrobial, antioxidant and cytotoxic activities of Anogeissus dhofarica from Oman. Int J Recent Adv Pharm Res. 2013;3:35–8.
27. Mann A, Amupitan J, Oyewale A, Okogun J, Ibrahim K. Chemistry of secondary metabolites and their antimicrobial activity in the drug development process: a review of the genus Anogeissus. Med Plants-Int J Phytomed Related Indust. 2009;1(2):55–77.
28. Marwah RG, Fatope MO, Al Mahrooqi R, Varma GB, Al Abadi H, Al-Burtamani SKS. Antioxidant capacity of some edible and wound healing plants in Oman. Food Chem. 2007;101(2):465–70.
29. Maqsood R, Khan F, Ullah S, Khan A, Al-Jahdhami H, Hussain J, et al. Evaluation of antiproliferative, antimicrobial, antioxidant, antidiabetic and phytochemical Analysis of Anogeissus dhofarica AJ Scott. Antibiotics. 2023;12(2);354.
30. Muanda FN, Bouayed J, Djilani A, Yao C, Soulimani R, Dicko A. Chemical composition and, cellular evaluation of the antioxidant activity of Desmodium adscendens leaves. Evid-Based Complement Altern Med. 2011;2011:9.
31. Luckett-Chastain L, Calhoun K, Schartz T, Gallucci RM. IL-6 influences the balance between M1 and M2 macrophages in a mouse model of irritant contact dermatitis. Am Assoc Immnol. 2016;196:196–7.
32. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186–95.