
INTRODUCTION
Age constitutes a nonmodifiable risk factor for a multitude of diseases, including Alzheimer’s disease and associated dementias [1], hypertension [2], type 2 diabetes mellitus [3], and cancer [4]. A systematic review reveals that multimorbidity affects a significant portion of the global population, particularly individuals aged 65 and older [5]. Medical research was previously focused on developing distinct treatments for these diseases. Understanding how aging can facilitate the development of chronic diseases, on the other hand, will enable the development of novel therapeutic approaches that target the aging process to treat numerous chronic diseases [6].
Aging itself is a complex process and there is no single molecular pathogenic pathway that can account for all features of aging [7]. However, accumulating evidence has shown that cellular senescence plays a significant role in many aging-related diseases [8,9]. Cellular senescence is a state of permanent cell-cycle arrest in response to various stressors such as oxidative stress, telomere shortening, DNA damage, and oncogene activation, or as part of physiological processes [10,11]. In senescent cells, cyclin-dependent kinase inhibitors (CDKIs) p21 and p16 are found in higher concentration, which hinders the progression of the cell cycle from the G1 to the S phase [11]. Although senescence is supposed to limit tumor formation, the presence of senescent cells is now known to promote cancers and other clinical diseases [12,13]. This is due to the release of pro-inflammatory cytokines, chemokines, and growth factors such as IL-6, IL-8, MIP-1, VEGF-1, and many others, collectively known as the senescence-associated secretory phenotype (SASP) [7,14]. Barinda et al. [15] found that parabiosis between wild-type mice and mice with senescent endothelium cells reduces the insulin sensitivity of the wild-type mice. As a blood-soluble factor, SASP mediates the systemic metabolic anomalies in wild-type mice [15]. Through the paracrine pathway, SASP can promote senescence in neighboring cells, creating a positive feedback loop [16].
Senescent cells are known to accumulate with aging. Senescence is hypothesized to influence aging via at least two separate mechanisms. First, homeostasis and regeneration capability are hampered by reduced cell proliferation brought on by increased production of anti-proliferative proteins such as CDKI. Second, the pro-inflammatory SASP will cause chronic inflammation and carcinogenesis, both of which are age-related symptoms [7].
Individual variations in the rate of aging may be accounted for in part by environmental agents known as gerontogens, which have the capacity to accelerate the molecular aging process. However, the current body of research on gerontogen remains scarce [6,7]. One of the challenges in identifying gerontogens is the lack of biomarkers to examine aging at the molecular level [7]. The measurement of senescence-related events, such as β-galactosidase staining, SASP levels in tissues, leukocyte telomere length, and p16 and p21 expression, is currently being investigated as a potential biomarker of aging [7,11]. To date, however, there has been no universal marker specifically expressed only by senescent cells. Even p16, which is considered more specific as a marker for senescence, was known to be expressed by certain nonsenescent cells and also not expressed by all cells that undergo senescence [17]. Other markers also have their shortcomings. Therefore, the selection of the right marker to detect senescence is also still a challenge.
Growing evidence showed that cellular senescence can be exploited as a therapeutic target to slow or prevent aging-related tissue dysfunction [9,18]. In mice, eliminating p16-expressing senescent cells slowed the emergence of aging-related clinical diseases such as cataracts [19]. The removal of senescent cells by prolonged administration of senolytic drugs can also alleviate vasomotor dysfunction in naturally aged and atherosclerotic mice. Elimination can also diminish osteogenesis markers in plaque, hence reducing plaque calcification in the mice tunica intima [20]. These investigations may help establish a link between senescence, aging, and the pathophysiology of chronic diseases that are common in older people and serve as a foundation for medication development to prevent or reverse aging.
This paper will discuss senescence inducers and biomarkers used in detecting senescence, recent research concerning gerontogens, and the ongoing development of senotherapy targeting senescent cells, with the aim of extending the human lifespan.
SENESCENCE INDUCERS
DNA damage
DNA damage, particularly DNA double-strand break (DSB), can cause senescence. This damage triggers the DNA damage response (DDR), a checkpoint that prevents cell cycle progression and genetic information loss in daughter cells. Proteins involved in DDR accumulate at the damage site and form foci due to chromatin modifications. These include phosphorylated histone H2AX and related proteins such as MDC1, 53BP1, and activated ataxia kinase mutated telangiectasia (ATM). These foci mark areas with DNA damage, leading to check-point and cell-cycle arrest until repair is achieved. If DNA damage persists, DDR signaling and cell cycle arrest continue, leading to senescence. At the end of the DDR signaling pathway, ATM activates p53, triggering the expression of CDKI p21, resulting in cell cycle termination [21].
Telomere shortening and damage
As cells replicate, the repeating regions at the end of linear chromosomes, the telomere, become shorter. When it is too short, the loss of protective structure in telomeres will be detected as DNA damage that is capable of activating DDR. Just like DNA damage, DDR activation activates p53 and causes the cell cycle to stop [21,22].
The mechanism by which telomere shortening can be identified as DNA damage is because the shortened telomere may no longer bind TRF2, one of the protective proteins that are present in telomeres, in adequate amounts. Van Steensel et al. [23] found that TRF2 gene mutations cause senescence. This is supported by in-vitro studies that showed TRF2 depletion in shortened telomeres can cause senescence [23]. The protective effect of TRF2 to prevent senescence was proved by Karlseder et al. [24] that found overexpression of TRF2 can protect short telomeres and lower the senescence setpoint from 7kb to 4kb. In contrast, the telomeres of nondividing cells, such as cardiomyocytes, adipocytes, neurons, osteocytes, and osteoblasts, do not shorten. Nevertheless, chronic activation of DDR can also occur when cells encounter DNA damage in the telomeres area, for instance, due to exposure to genotoxic substances [21].
Oncogene activation
Oncogene activation and tumor suppressor gene inactivation will activate the p53/p21 and p16 pathways [25]. Initial activation of oncogenes induces a hyperproliferative phase that is fundamentally linked to the interruption of DNA replication that triggers DDR activation [21]. Bartkova et al. [26] demonstrate that replicative stress in oncogene-induced senescence induces premature termination of the replication fork, resulting in DNA DSBs [26].
Mitochondrial dysfunction
Mitochondrial dysfunction can lead to the occurrence of senescence through the activation of AMPK and the p53 pathway. Mitochondria converts NADH to NAD+, so a decrease in NAD+/NADH ratio indicates mitochondrial dysfunction. NAD+/NADH ratio regulates energy production because NAD+ is necessary for glycolysis while NADH inhibits it. When the ratio of NAD+ to NADH falls, as an energy sensor, AMPK is activated [27]. Senescence caused by mitochondrial dysfunction is referred to as mitochondrial dysfunction-induced senescence and exhibits a distinct set of SASP compared to other types of senescence [13].
BIOMARKERS OF SENESCENCE
Senescence biomarkers can be used to identify aging due to the close relationship between aging and senescence. Two or more senescent cell markers are recommended for detecting senescence [28–30]. Gonzalez-Gualda et al. [28] advocate utilizing three types of markers to indicate three different processes: cell-cycle arrest, structural changes, and markers unique to the subtypes of senescence to be researched, such as DNA damage, SASP factors, or ROS levels [28]. Kohli et al. [30] suggest a two-phase senescence marker measurement. Senescence is validated using universal markers in the first step, whereas subtypes are identified in the second phase [30]. To date, however, the selection of markers has been left to the discretion of the researcher [29].
Senescence markers are observable both in vitro and in vivo/ex vivo. However, there are significant limits to in-vivo detections. In contrast to cell culture, the majority of organismal cells experience quiescence or are already terminally differentiated. Quiescence is a state of transient cell cycle arrest. In contrast to senescence, quiescent cells are capable of re-entering the cell cycle when triggered by the appropriate mitogen [7]. Meanwhile, cells that have reached terminal differentiation will perform specified activities and cease to proliferate [11]. Therefore, markers related to cell growth and DNA replication become invalid for the in-vivo detection of senescence. Moreover, changes in morphology or an increase in cell size that can be noticed in vitro are typically not preserved in vivo due to structural and architectural constraints. Cell cycle-associated proteins can also be expressed by nonsenescent cells and certain processes such as inflammation, so the reliability of these markers is also limited [28].
Markers for cell-cycle arrest, cell proliferation, and DNA replication
Biomarkers related to cellular senescence have been summarized in Table 1. The cell-cycle arrest is a key hallmark of senescence, marked by increased p16, p21, and p53 proteins and decreased phosphorylated retinoblastoma protein. In senescence, the p16/Rb and p53/p21 axes cause cell-cycle arrest. After both processes, hypophosphorylated Rb interacts with the E2F transcription factor. E2F induces cell cycle progression and S phase. When attached to Rb, E2F cannot interact with its target gene’s promoter, preventing gene transcription needed for replication and stopping the cell cycle [28].
On the p16/Rb axis, senescence-inducing stimuli will activate the INK4a/ARF locus, hence increasing p16 levels. The p16 protein, which is a CDKI, inhibits the development of the CDK4-Cyclin D complex, resulting in the dephosphorylation and stability of the Rb-E2F complex and the cessation of the cell cycle. Typically, the p16/Rb axis is activated during senescence produced by replicative stress, reactive oxygen species, and oncogene activation. It is believed that the p16/Rb pathway has a more critical role in sustaining senescence. Activation of the p53/p21 axis begins with the phosphorylation of p53 (p-p53), which induces the overexpression of the CDKI p21. p21 inhibits the formation of the CDK2-Cyclin E complex, which is responsible for Rb dephosphorylation and E2F sequestration, hence halting the cell cycle. In senescence triggered by replicative stress, DNA damage, reactive oxygen species, and oncogene activation, the p53/p21 axis becomes activated. This route is hypothesized to be activated during the early stages of senescence [28].
Positive ex-vivo results should be interpreted with caution because p16 is also expressed by aged lymphocytes, cells that undergo Rb inactivation (e.g., tumor cells), and certain physiological circumstances such as inflammation, clastogen exposure, and wound healing. Currently, available antibodies for histochemical detection of p16 are likewise unreliable. To address this, researchers have designed experimental mice containing a reporter gene on the p16 promoter. In addition to p16, the use of the marker p53/p21 ex vivo is limited since transient damage can activate p53 for the DNA repair process in quiescent cells and also plays a role in the apoptosis process [28].
The cell-cycle arrest also needs to be evaluated through examination of cell proliferation and/or DNA replication. Cell proliferation can be measured by performing cell counting over time to obtain cell growth profiles in vitro [28]. Another proliferative marker is the Ki-67 protein which can be examined through immunostaining. Ki-67 is a nuclear protein that can be found in all active phases of the cell cycle or as cells proliferate [28,31]. However, the use of proliferative markers such as Ki-67 in living organisms has limitations because most cells are in a state of quiescence or have reached final differentiation [28].
DNA synthesis examination can be done by incorporating modified nucleotides, such as 5-ethynyl-2’-deoxyuridine (EdU) or 5-bromo-2-deoxyuridine (BrdU), into the DNA of replicating cells. The cell cycle arrest is defined by a reduction of replicative ability, which means that fewer nucleotides will be integrated throughout the replication process. Fluorescent imaging or flow cytometry analyses are utilized to detect these nucleotides [28].
Senescence-associated β-galactosidase (SA-β-Gal)
β-Gal is a hydrolase enzyme found in lysosomes that catalyzes the hydrolysis of β-galactosidase into monosaccharides. The majority of cells have endogenous β-galactosidase, which is active at pH 4.0. When the pH of lysosomes is elevated to 6, the activity of β-galactosidase expressed by nonsenescent cells is reduced, while the activity of β-Gal expressed by senescent cells is increased. To discriminate senescent cells from nonsenescent cells, the analysis of β-Gal associated with senescence (SA-β-Gal) is conducted at a suboptimal pH (6.0) [28,32].
SA-β-Gal is one of the most commonly employed biomarkers for histological or cytological detection of senescence. SA-β-Gal can be examined in vitro by adding the chromogenic substrate 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (XGal) which forms a blue precipitate. Additionally, SA-β-Gal testing can be performed on living organisms by using galactose coupled to a fluorescence probe, such as SPiDER-BGal. This probe will be “activated” when the sugar it contains is degraded by B-galactosidase, allowing it to be used to mark senescent cells with elevated B-gal activity, which may subsequently be detected using flow cytometry techniques. However, just like other markers, β-Gal is not only expressed by senescent cells in living organisms, but is also found in secretory cells such as tissue macrophages and osteoclasts, cells that experience increased lysosomal activity during the autophagy process, and some cancer cells [28].
Nuclear alteration
Heterochromatin is sparsely distributed at the periphery of the nucleus in nonsenescent dividing cells. However, in senescent cells, the chromatin condensed and is known as senescence-associated heterochromatin foci (SAHF). Chromatin condensation occurs to silence the genes needed for the proliferation process. SAHF examination can be performed by staining using 40,6-diamidino-2-phenylindole and confocal microscopy imaging techniques. In addition, SAHF also contains heterochromatin-forming proteins, such as HP1, H3K9me, and H2A macro histones that can be examined by immunostaining [28].
Another more global marker to detect senescence is DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence). DNA-SCARS is a nuclear structure that contains various proteins related to DDR. [28].
Telomere length has also been used to detect senescence. Telomere length can be measured by qPCR technique and quantitative fluorescent in situ hybridization. In addition to telomere length, the formation of telomere dysfunction-induced foci (TIFs) can also be used as a senescence marker. TIFs are DNA-SCARS formed in telomeres. In other words, DNA damage markers, such as γ-H2AX, ATM, and 53Bp1 are found in telomere regions. TIF can be found on telomeres that do not experience shortening, so TIF can also be found in various types of senescence, not just those caused by replicative stress. The detection method for TIF is generally the same as DNA-SCARS because the components are similar [28].
Lamin B1 protein is downregulated in senescent cells. Lamin B1, a significant nuclear lamina component, maintains nuclear structural integrity. In senescent cells, downregulation of lamin B1 allows chromatin fragments to escape from the nucleus. Immunoassay or immunoblotting can be used to detect lamin B1 depletion [28].
Senescence-associated secretory phenotype
SASP is a set of factors, including cytokines, chemokines, growth factors, metalloproteinases, and extracellular vesicles secreted by senescent. SASP can be examined through RT-qPCR, proteomic techniques (LC-MS/MS), and ELISA [28].
The majority of research that correlates senescence with age-related diseases in human tissues does not employ SASP as a marker [29]. Many of these substances are also released by other cells, including immune cells, endothelium, and cancer cells, therefore SASP cannot be used alone as a reliable indicator of senescence. In addition, its expression fluctuated and evolved. The SASP profile depends on the duration, type of stressor, and cell type [28]. SASP from distinct cells within the same tissue exhibit distinct characteristics. SASP is also known to trigger senescence in neighboring cells; therefore, the senescence marker can change as senescence occurs in the network of neighboring cells [29].
To use SASP as a more specific marker, it is necessary to first determine the SASP profile of the tissue/organ under research. Chatsirisupachai’s transcriptome studies revealed that gene patterns in various senescent tissues are distinct. The outcomes of these investigations can serve as a basis for selecting the appropriate SASP markers for specific tissues [29]. In addition, the SASP Atlas database provides several details regarding dissolved protein components and exosome contents from fibroblast cells and endothelial cells that undergo senescence following oncogene activation, radiation, and specific treatment. The SASP Atlas demonstrates that each stressor has a unique SASP profile and that the overlap is extremely limited, making it difficult to identify a universal SASP marker [28].
Mitochondria, reactive oxygen species (ROS) and pro-survival pathways
In senescent cells, mitochondria enlarge, and their number increases due to the decreased mitophagy process. Mitochondria also play a role in causing an increase in ROS which is also a marker of senescence. Mitochondrial ROS released by senescent cells may cause senescence by autocrine and paracrine mechanisms. ROS can be detected by various stains, e.g., Dihydroethidium (DHE). DHE that is blue will undergo oxidation by intracellular superoxide and turn red [28].
Another hallmark of senescence is apoptosis resistance. Apoptosis resistance is caused by Bcl-2 overexpression in mitochondrial membranes and endoplasmic reticulum, which activates pro-survival signaling pathways. Bcl-2 inhibits the release of cytochrome c and the subsequent activation of a caspase signaling pathway that triggers intrinsic apoptosis. A more common method employed in the detection of apoptosis resistance is by excluding apoptosis, marked by the absence of annexin V, cleaved caspase 3, and PARP [28].
GERONTOGEN
We have undoubtedly observed that some people live longer than others and have contemplated the reasons behind this phenomenon. Various research found that longevity is influenced by both genetic and environmental factors. This can be gleaned from research findings on the factors impacting longevity among centenarians (individuals aged 99 years and older), including studies conducted in Japan. Okinawa is one such region in Japan that exhibits a higher prevalence of centenarians compared to other relatively long-lived countries and regions. Beyond simply reaching an advanced age, Okinawan centenarians also appear to maintain functional independence well into their 90s [33,34].
Research on centenarians in Okinawa indicates that among the factors contributing to how the Okinawans achieve a very long lifespan are lifestyle and genetics. Until the 1960s, when centenarians were still in their middle age, the dietary pattern of Okinawans resembled that of a naturally calorically restricted population. Okinawans were also physically active, with a BMI around 21, and experienced fewer age-related diseases. The lifestyle changes occurring among younger generations of Okinawans, such as higher-calorie diets, reduced physical activity, and other risky behaviors, have resulted in a higher prevalence of lifestyle-related diseases such as diabetes and obesity [33].
The study on centenarians in Okinawa also indicates the role of genetics in longevity, where more longevity was found among siblings in centenarian families compared to control families. One of the genes implicated in longevity is FOXO3, which has recently been discovered, may exert its effects through various pro-longevity mechanisms, the nature of which may vary depending on age and sex [33,35].
 | Table 1. Biomarkers of senescence [28]. [Click here to view] |
When comparing the factors that have a greater impact on longevity, the answer lies in environmental factors, as a recent study has reported that the true heritability of human longevity was concluded to be well below 10% [36]. Currently, there exists a term known as gerontogen, which refers to environmental factors or agents that can accelerate molecular aging, consequently affecting longevity [7]. Up to this point, several gerontogens have already been identified, including UV exposure, high-fat diet, heavy metal exposure, cigarette smoke, and psychological stress coming from adverse social environments, which will be discussed individually in this subsection.
UV radiation
The sun emits UVA (320–400 nm), UVB (280–320), and UVC rays (100–280 nm). UVC is absorbed by the stratosphere, allowing only ~95% of UVA and ~5% of UVB to reach Earth’s surface. Reactive oxygen species generated by UVA and UVB can cause indirect damage to the DNA. In addition, UVB can directly damage the DNA through the formation of a cyclobutene pyrimidine dimer [37]. Sorrentino et al. [45] found that continuous exposure to UVB can lead to an increase in DNA damage markers, including phosphorylated-H2AX, p16, Cxcl1, and IL-6.15. According to Ma et al. [38], in-vitro UVA radiation causes senescence in human fibroblast cells through telomeres shortening.
High-fat diet
Kim et al. [39] show that administration of a high-fat diet (60% fat) for 6 months in mice can cause impaired kidney function and elevated senescence markers in the kidneys, which include SA-β-Gal, p16, p19, and p53 as well as IL-1α, MCP1, and TNF-α. Meanwhile, p21 did not experience a significant increase. Senescent cell elimination with quercetin improved kidney function. High-fat diets can promote dyslipidemia and obesity, which may contribute to senescence [39]. Obesity is associated with chronic inflammatory conditions and increased oxidative stress. Chronic inflammation is known to accelerate the aging process. In the absence of any other genetic or environmental factor, Jurk et al. [40] discovered that systemic chronic inflammation can accelerate aging via ROS-mediated exacerbation of telomere dysfunction and cellular senescence. In addition, obese individuals have shorter telomeres [41–43] and a diminished ability to counteract oxidative damage [44]. Another study conducted by Sorrentino et al. [45] using p16-reporter mice showed that a high-fat diet (42% fat) did not increase the expression of p16 assessed by Total-Body Luciferase Imaging (TBLI). This may be due to senescence that occurs on a scale that cannot yet be detected by TBLI measurements [45].
Heavy metal
Currently, there has been a review of heavy metals that have been shown to induce senescence, both in vitro and in vivo, including arsenic and iron [46]. Arsenic (As) is a toxicant commonly found in air, water, and soil and can enter the body through various routes. Exposure to arsenic can increase ROS production which causes DNA damage and affects the DNA repair process [7,45,46]. Okamura et al. [47] showed that exposure to sodium arsenite 5 and 7.5 μg for 144 hours caused the occurrence of senescence in the human stellate cell line, which is characterized by changes in cell morphology, increased SA-β-Gal, increased expression of p21, increased marker of DNA damage (γ-H2AX), and decreased expression of lamin B1. In addition, there is upregulation of various SASP mRNAs, including MMP1, MMP3, IL-8, IL-1β, and Cxcl1. The increase in DNA damage markers indicates the role of DNA damage in causing senescence as a result of arsenic exposure [47]. In-vivo research by Sorrentino et al. [45] showed that chronic exposure to arsenic as much as 50 ppm through drinking water led to increased p16 expression in mice detected at week 24 [45].
Meanwhile, iron (Fe), in the form of iron oxide, is found as air pollutants from exhaust emissions [48]. Excess iron in the body can lead to the formation of ROS through the Fenton and Haber-Weiss reactions [49]. Noh et al. [50] show that iron overload may cause senescence in cerebral endothelial cells derived from aging mice, especially in cells derived from female mice. In that research, senescence is characterized by a decrease in cell proliferation that is not accompanied by cell death, an increase in DNA damage markers (γ-H2AX), and the expression of p16, p21, and IL-6. There is a reciprocal relationship between iron and senescence in which senescent cells show iron accumulation, and iron accumulation causes senescence [50].
Cigarette smoke
Senescence is related to the occurrence of several lung pathologies, such as fibrotic pulmonary disease [51] and chronic obstructive pulmonary disease [52]. One of the factors that cause senescence in the lungs is cigarette smoke [53]. Cigarette smoke contains various mutagens, such as formaldehyde, carbon monoxide, and nicotine that can cause DNA damage [7]. Nicotine exposure, according to Bodas et al. [54] causes bronchial epithelial cell senescence via ROS-mediated autophagy impairment. Meanwhile, in an experiment conducted by Sorrentino et al. [45] mice exposed to cigarette smoke showed a significant increase in p16 after a few weeks and remained persistent even after exposure was stopped Table 2.
Adverse social environment
In their publication, Sorrentino et al. [45] classified psychological stress as a distinct class in its role as a gerontogen [7]. Life stress, particularly arising from adverse social environments, has indeed been demonstrated to trigger hallmarks of aging, including cellular senescence. Research by Rentscher et al. [55] indicates that mid-life parents who reported greater exposure to chronic psychological stress, perceived stress, and accumulated daily stress appraisals exhibited increased expression of p16. Carroll et al. [56] demonstrated that low social support (defined as the availability of someone who will listen, give advice, show affection/love, and provide emotional support) in late life is associated with telomere shortening. Lin et al. [57] conducted a study using a chronic unpredictable stress (CUS) animal model intended to resemble stressful life conditions. The results of this study indicated that CUS-treated mice exhibited depression-like behavior and cognitive decline, along with increased expression of the protein p16 in the hippocampus and senescence-associated β-galactosidase (SA-β-gal) in the hippocampal dentate gyrus. The elevated markers of senescence in this study also strongly correlated with the severity of memory impairment observed in CUS-treated mice. These findings suggest a role for stress in triggering cellular senescence, subsequently leading to cognitive decline [57].
The biological changes occurring with the aging process triggered by the social environment can, in turn, have an impact on an individual’s social life. For instance, as reviewed by Siracusa et al. [58] cognitive decline in the form of memory impairment can affect an individual’s ability to maintain friendships and engage in social activities. Declines in hippocampal function can also affect an individual’s ability to process information, which subsequently impacts their ability to attend to and respond to social cues and adjust their behavior accordingly. Ultimately, this can lead to increased social withdrawal [58].
SENOTHERAPY AS AN APPROACH TO DELAY AGING AND EXTENDS HUMAN LIFESPAN
Currently, Jeanne Calment, who died at the age of 122, remains the oldest documented human individual ever lived [59]. However, discussions regarding the maximum lifespan for humans continue to be debated among researchers. In their study, Dong et al. [59] claimed to have found evidence of a natural limit to human lifespan. Dong et al. [59] conducted demographic analysis using yearly maximum reported age at death (MRAD) data from 1968 to 2006. They then divided the data into two groups (1968-1994 and 1995-2006) and found that before 1995, MRAD increased by 0.15 years per year, but after 1995, MRAD decreased by 0.28 years per year. The data shows that human yearly MRAD has plateaued at the age of 114 years. They then estimated that the likelihood of MRAD exceeding 125 years is very small [59]. However, Dong et al.’s [59] findings have been criticized by some researchers regarding the methodology used, leading to questions about the conclusions drawn from the study and deemed unable to prove the existence of a limit to human lifespan [60–63]. Meanwhile, de Beer et al. [62] argue that the limit to human lifespan around 115 years predicated upon the unchanged MRAD post-1995 as evidenced in Dong et al.’s [59] study does not mean that someone cannot live beyond the age of 115 in the future. They then predict that the maximum human lifespan could reach 125 years [62]. Regardless of the presence or absence of defined boundaries, efforts have been devoted to slowing down aging processes and extending human lifespan. The close connection between senescence and aging with the pathophysiology of chronic diseases, characterized by shared hallmarks (as illustrated in Table 3) has sparked interest in using senescent cells as therapeutic targets. As demonstrated by various research studies, therapeutically targeting senescence (senotherapy) has been shown to extend lifespan, at least in animal models [64]. Senotherapy, which is divided into senolytic and senomorphic agents, will be discussed below.
Senolytic and senomorphic agents
Currently, medicines known as senolytic agents have been developed to eliminate senescent cells in various ways. For example, navitoclax drugs (ABT-737 and ABT-263) work by inhibiting the activity of Bcl-2 so that senescent cells can be removed through apoptosis. Other drugs use the overexpression of SA-β-gal as a target. Due to the enhanced activity of SA-β-gal in senescent cells, nanoparticles coated with galacto-oligosaccharides can target senescent cells more precisely [21].
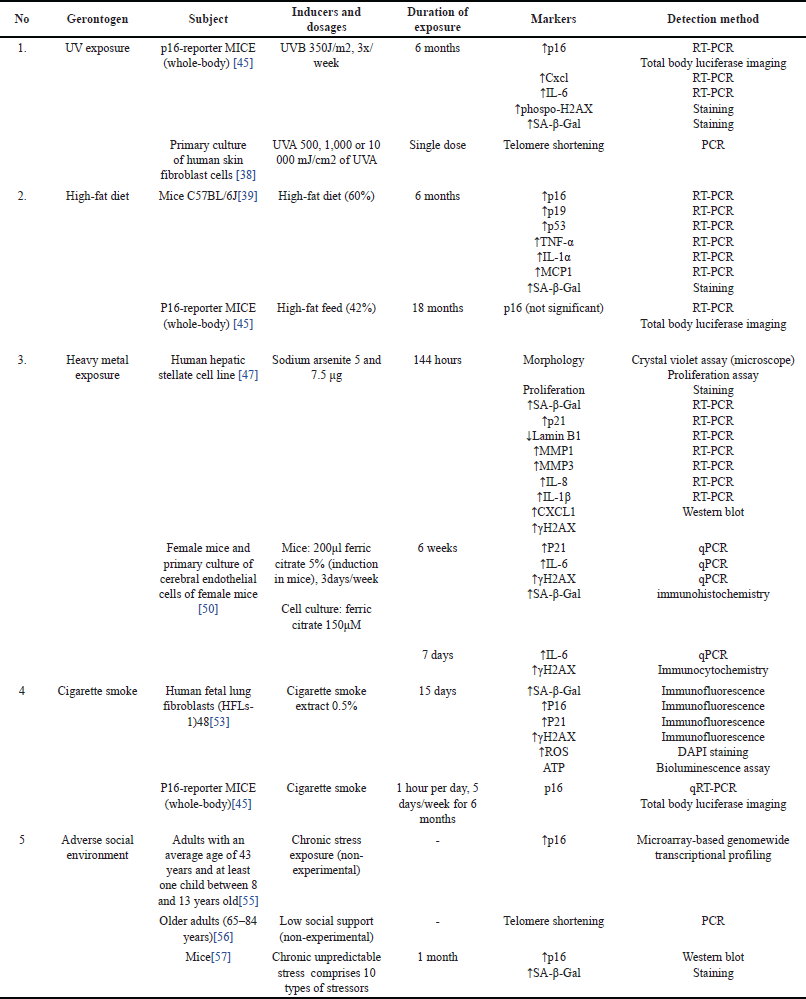 | Table 2. Gerontogen and the corresponding alterations in senescence biomarkers. [Click here to view] |
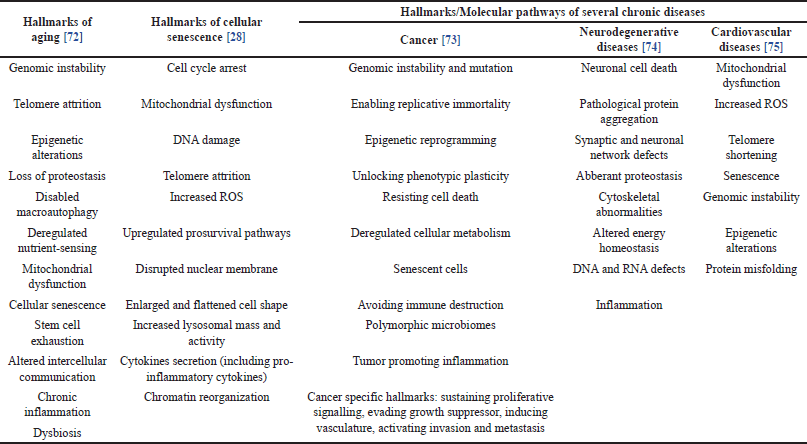 | Table 3. Hallmarks of aging, cellular senescence, and several chronic diseases. [Click here to view] |
The combination of dasatinib with quercetin (D+Q) constitutes another senolytic agent. Dasatinib works by targeting the senescence cell antiapoptotic pathway through tyrosine kinase inhibition, while quercetin is a natural flavonoid that works by inhibiting BCL-2/BCL-XL, PI3K/AKT, and p53/p21 [65,66]. In experimental animal models, D+Q was shown to reduce the incidence of intervertebral disc degeneration associated with the aging process accompanied by a decrease in senescence markers, including p16, p19, and SASP (IL-6 and MMP13) in mice [65].
In clinical trials, a pilot study in phase I clinical trials showed that oral administration of D+Q combination for 3 days could eliminate senescent cells in adipose tissue of diabetic kidney injury patients on the 11th day after therapy. In the study, there was a decrease in the number of cells expressing p16 and p21, cells with increased SA-β-Gal, and SASP (IL-1α, IL-6, MMP9, and MMP12) [67]. The use of DQ in idiopathic pulmonary fibrosis (IPF) patients has also been evaluated in the first in-human clinical trial in the United States. In the study, oral DQ (dasatinib 100 mg/day and quercetin 5 × 150 mg/day) was given intermittently for 3 weeks, where each week, DQ was given 3 days in a row, followed by a 4-day no-drug period (total 9 doses). The results showed that intermittent DQ administration for 3 weeks produced good adherence and could improve the physical function of IPF patients. From the aspect of safety and tolerability, in general, adverse events that occur in the administration of the regimen are classified as mild-moderate, reversible, and do not cause clinically relevant sequelae [68].
Senomorphic agents serve as an alternative to senolytic agents. The mode of action of a senomorphic agent is to alter various properties of senescent cells, particularly those associated with the generation and secretion of SASP while preserving the viability of the cell. This method will diminish the pro-inflammatory effects caused by senescent cells. Compounds that affect NF-B signaling, including metformin, apigenin, and kaempferol, are known to reduce SASP synthesis. In addition, other neutralizing antibodies for SASP or its receptors, such as IL-6, IL-1, IL-1, and TNF, had senomorphic effects [21].
Currently, there is also a review regarding the anti-senescence properties of various Indonesian natural ingredients [69]. Turmeric (Curcuma longa) containing curcumin has antioxidant effects and exhibits senolytic effects in mice. The content of piperlongumine in Cabe Jawa (Piper retrofractum Vahl.) is known to have a senolytic effect by inducing apoptosis in senescent fibroblast cells. Pegagan (Centella asiatica) can prevent senescence in fibroblast cells through its antioxidant effects [69].
The use of senolytic and senomorphic agents has its own advantages and disadvantages. For senomorphic agents that work by inhibiting SASP, continuous treatment is needed to maintain SASP suppression and this can cause side effects and off-target effects due to the suppression of cytokine secretion by nonsenescent cells, such as innate and adaptive immune cells. Meanwhile, senolytic agents that work by targeting the underlying cause of SASP by eliminating senescent cells, can be administered intermittently and their effectiveness is equivalent to continuous administration. This “hit-and-run” strategy in the administration of senolytic agents can reduce side effects [70]. Nevertheless, complete eradication of senescent cells can also be dangerous since senescence is also needed in some important processes such as wound healing. This is also applicable to the inhibition of regulatory pathways, including NF-κB, which controls inflammation and the immune response in addition to being involved in SASP secretion. Therefore, the development of senotherapy must also consider other beneficial functions of the system to be targeted [70,71].
CONCLUSION
In this review, we emphasize the role of environmental factors in accelerating the aging process through discussions on gerontogens. Environmental factors, which significantly contribute to aging and longevity, are modifiable factors. We contend that interventions at both individual and governmental levels, focusing on healthy lifestyles and environmental enhancements, can facilitate healthy aging and lifespan extension. Understanding the impact of aging on individuals’ physical and psychological well-being, alongside the recognition that biological aging may manifest more rapidly in certain individuals, should foster greater understanding and empathy toward the challenges faced not only by chronologically older individuals but also by those who are biologically older.
We also highlight the increasing interest in using senescent cells as a therapeutic target for the prevention or treatment of numerous diseases. However, the lack of a common marker for senescence makes identification challenging, thus limiting gerontogen research. The challenge lies in developing a detectable marker for identifying unlimited gerontogens to enhance the understanding of aging and chronic diseases induced by environmental factors. Even though this limitation also has an impact on the senotherapy research, numerous studies remain ongoing and have yielded a number of promising agents. While the preliminary findings appear favorable, the advancement of senotherapy, particularly senolytics, still necessitates the investigation and validation of a number of critical factors, including the potential for off-target effects and specificity, which must be addressed to avoid possible hazardous side effects.
ACKNOWLEDGMENTS
The first author wishes to express gratitude to the Indonesia Endowment Fund for Education Agency (LPDP) for their financial support towards the publication of this paper.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVAL
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER'S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Litke R, Garcharna LC, Jiwani S, Neugroschl J. Modifiable risk factors in Alzheimer disease and related dementias: a review. Clin Ther. 2021;43(6):953–65. CrossRef
2. Wu J, Li T, Song X, Sun W, Zhang Y, Liu Y, et al. Prevalence and distribution of hypertension and related risk factors in Jilin Province, China 2015: a cross-sectional study. BMJ Open. 2018;8(3):e020126. CrossRef
3. Nguyen CT, Pham NM, Lee AH, Binns CW. Prevalence of and risk factors for type 2 diabetes mellitus in Vietnam: a systematic review. Asia Pac J Public Health. 2015;27(6):588–600. CrossRef
4. Arafat HM, Omar J, Muhamad R, Al-Astani TAD, Shafii N, Al Laham NA, et al. Breast cancer risk from modifiable and non-modifiable risk factors among palestinian women: a systematic review and meta-analysis. Asian Pac J Cancer Prev. 2021;22(7):1987–95. CrossRef
5. Nguyen H, Manolova G, Daskalopoulou C, Vitoratou S, Prince M, Prina AM. Prevalence of multimorbidity in community settings: a systematic review and meta-analysis of observational studies. J Comorb. 2019;9:2235042X1987093. CrossRef
6. Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159(4):709–13. CrossRef
7. Sorrentino JA, Sanoff HK, Sharpless NE. Defining the toxicology of aging. Trends Mol Med. 2014;20(7):375–84. CrossRef
8. Salam R, Saliou A, Bielle F, Bertrand M, Antoniewski C, Carpentier C, et al. Cellular senescence in malignant cells promotes tumor progression in mouse and patient Glioblastoma. Nat Commun. 2023;14(1):441. CrossRef
9. Matsudaira T, Nakano S, Konishi Y, Kawamoto S, Uemura K, Kondo T, et al. Cellular senescence in white matter microglia is induced during ageing in mice and exacerbates the neuroinflammatory phenotype. Commun Biol. 2023;6(1):665. CrossRef
10. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–27. CrossRef
11. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging. 2021;1(10):870–9. CrossRef
12. Mario Gonzalez-Meljem J, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017;8(1):1819. CrossRef
13. Dou X, Fu Q, Long Q, Liu S, Zou Y, Fu D, et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat Metab. Published online November 1, 2023;5(11):1887–910. CrossRef
14. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. CrossRef
15. Barinda AJ, Ikeda K, Nugroho DB, Wardhana DA, Sasaki N, Honda S, et al. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat Commun. 2020;11(1):1–13. CrossRef
16. Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes. 2015;64(7):2289–98. CrossRef
17. Frescas D, Hall BM, Strom E, Virtuoso LP, Gupta M, Gleiberman AS, et al. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle. 2017;16(16):1526–33. CrossRef
18. Zonari A, Brace LE, Al-Katib K, Porto WF, Foyt D, Guiang M, et al. Senotherapeutic peptide treatment reduces biological age and senescence burden in human skin models. npj Aging. 2023;9(1):10. CrossRef
19. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–6. CrossRef
20. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–7. CrossRef
21. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22(2):75–95. CrossRef
22. Rossiello F, Jurk D, Passos JF, d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24(2):135–47. CrossRef
23. van Steensel B, Smogorzewska A, de Lange T. TRF2 Protects human telomeres from end-to-end fusions. Cell. 1998;92(3):401–13. CrossRef
24. Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science (1979). 2002;295(5564):2446–9. CrossRef
25. Domen A, Deben C, Verswyvel J, Flieswasser T, Prenen H, Peeters M, et al. Cellular senescence in cancer : clinical detection and prognostic implications. J Exp Clin Cancer Res. Published online 2022:41(1):360. CrossRef
26. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–7. CrossRef
27. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–14. CrossRef
28. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288(1):56–80. CrossRef
29. Tuttle CSL, Luesken SWM, Waaijer MEC, Maier AB. Senescence in tissue samples of humans with age-related diseases: a systematic review. Ageing Res Rev. 2021;68(March):101334. CrossRef
30. Kohli J, Wang B, Brandenburg SM, Basisty N, Evangelou K, Varela-Eirin M, et al. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat Protoc. 2021;16:2471–98; CrossRef
31. Uxa S, Castillo-Binder P, Kohler R, Stangner K, Müller GA, Engeland K. Ki-67 gene expression. Cell Death Differ. 2021;28(12):3357–70. CrossRef
32. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated β-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(20):3613–22. CrossRef
33. Willcox BJ, Willcox DC, Suzuki M. Demographic, phenotypic, and genetic characteristics of centenarians in Okinawa and Japan: Part 1—centenarians in Okinawa. Mech Ageing Dev. 2017;165:75–9. CrossRef
34. Willcox DC, Willcox BJ, Shimajiri S, Kurechi S, Suzuki M. Aging gracefully: a retrospective analysis of functional status in okinawan centenarians. Am J Geriatr Psychiatry. 2007;15(3):252–6. CrossRef
35. Torigoe TH, Willcox DC, Shimabukuro M, Higa M, Gerschenson M, Andrukhiv A, et al. Novel protective effect of the FOXO3 longevity genotype on mechanisms of cellular aging in Okinawans. npj Aging. 2024;10(1):18. CrossRef
36. Graham Ruby J, Wright KM, Rand KA, Kermany A, Noto K, Curtis D, et al. Estimates of the heritability of human longevity are substantially inflated due to assortative mating. Genetics. 2018;210(3):1109–24. CrossRef
37. Fitsiou E, Pulido T, Campisi J, Alimirah F, Demaria M. Cellular senescence and the senescence-associated secretory phenotype as drivers of skin photoaging. J Investig Dermatol. 2021;141(4):1119–26. CrossRef
38. Ma HM, Liu W, Zhang P, Yuan XY. Human skin fibroblast telomeres are shortened after ultraviolet irradiation. J Int Med Res. 2012;40(5):1871–7. CrossRef
39. Kim SR, Jiang K, Ogrodnik M, Chen X, Zhu XY, Lohmeier H, et al. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res. 2019;213:112–23. CrossRef
40. Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172. CrossRef
41. Moreno-Navarrete JM, Ortega F, Sabater M, Ricart W, Fernández-Real JM. Telomere length of subcutaneous adipose tissue cells is shorter in obese and formerly obese subjects. Int J Obes. 2010;34(8):1345–8. CrossRef
42. Clemente DBP, Maitre L, Bustamante M, Chatzi L, Roumeliotaki T, Fossati S, et al. Obesity is associated with shorter telomeres in 8 year-old children. Sci Rep. 2019;9(1):18739. CrossRef
43. Batsis JA, Mackenzie TA, Vasquez E, Germain CM, Emeny RT, Rippberger P, et al. Association of adiposity, telomere length and mortality: data from the NHANES 1999-2002. Int J Obes. 2018;42(2):198–204. CrossRef
44. Grun LK, Teixeira N da R, Mengden L von, de Bastiani MA, Parisi MM, Bortolin R, et al. TRF1 as a major contributor for telomeres’ shortening in the context of obesity. Free Radic Biol Med. 2018;129(September):286–95. CrossRef
45. Sorrentino JA, Krishnamurthy J, Tilley S, Alb JG, Burd CE, Sharpless NE. P16INK4a reporter mice reveal age-promoting effects of environmental toxicants. J Clin Investig. 2014;124(1):169–73. CrossRef
46. Vielee ST, Wise JP. Among gerontogens, heavy metals are a class of their own: a review of the evidence for cellular senescence. Brain Sci. 2023;13(3):500. CrossRef
47. Okamura K, Sato M, Suzuki T, Nohara K. Inorganic arsenic exposure-induced premature senescence and senescence-associated secretory phenotype (SASP) in human hepatic stellate cells1. Toxicol Appl Pharmacol. 2022;454(September):116231. CrossRef
48. Maher BA, González-Maciel A, Reynoso-Robles R, Torres-Jardón R, Calderón-Garcidueñas L. Iron-rich air pollution nanoparticles: an unrecognised environmental risk factor for myocardial mitochondrial dysfunction and cardiac oxidative stress. Environ Res. 2020;188:109816. CrossRef
49. Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. 2019;1863(9):1398–409. CrossRef
50. Noh B, Blasco-Conesa MP, Rahman SM, Monga S, Ritzel R, Guzman G, et al. Iron overload induces cerebral endothelial senescence in aged mice and in primary culture in a sex- dependent manner. Aging Cell. 2023 Nov;22(11):e13977. CrossRef
51. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. CrossRef
52. Zeng M, Zhang X, Xing W, Wang Q, Liang G, He Z. Cigarette smoke extract mediates cell premature senescence in chronic obstructive pulmonary disease patients by up-regulating USP7 to activate p300-p53/p21 pathway. Toxicol Lett. 2022;359:31–45. CrossRef
53. Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 2015;29(7):2912–29. CrossRef
54. Bodas M, Van Westphal C, Carpenter-Thompson R, Mohanty DK, Vij N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic Biol Med. 2016;97:441–53. CrossRef
55. Rentscher KE, Carroll JE, Repetti RL, Cole SW, Reynolds BM, Robles TF. Chronic stress exposure and daily stress appraisals relate to biological aging marker p16 INK4a. Psychoneuroendocrinology. 2019;102:139–48. CrossRef
56. Carroll JE, Diez Roux A V, Fitzpatrick AL, Seeman T. Low social support is associated with shorter leukocyte telomere length in late life: multi-ethnic study of atherosclerosis. Psychosom Med. 2013;75(2):171–7. CrossRef
57. Lin YF, Wang LY, Chen CS, Li CC, Hsiao YH. Cellular senescence as a driver of cognitive decline triggered by chronic unpredictable stress. Neurobiol Stress. 2021;15:100341. CrossRef
58. Siracusa ER, Higham JP, Snyder-Mackler N, Brent LJN. Social ageing: exploring the drivers of late-life changes in social behaviour in mammals. Biol Lett. 2022;18(3):20210643. CrossRef
59. Dong X, Milholland B, Vijg J. Evidence for a limit to human lifespan. Nature. 2016;538(7624):257–9. CrossRef
60. Lenart A, Vaupel JW. Questionable evidence for a limit to human lifespan. Nature. 2017;546(7660):E13–4. CrossRef
61. Rozing MP, Kirkwood TBL, Westendorp RGJ. Is there evidence for a limit to human lifespan? Nature. 2017;546(7660):E11–2. CrossRef
62. de Beer J, Bardoutsos A, Janssen F. Maximum human lifespan may increase to 125 years. Nature. 2017;546(7660):E16–7. CrossRef
63. Brown NJL, Albers CJ, Ritchie SJ. Contesting the evidence for limited human lifespan. Nature. 2017;546(7660):E6–E7. CrossRef
64. Suda M, Shimizu I, Katsuumi G, Yoshida Y, Hayashi Y, Ikegami R, et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat Aging. 2021;1(12):1117–26. CrossRef
65. Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA, et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneraation in mice. Nat Commun. 2021;12(1):5213. CrossRef
66. Van houcke J, Mariën V, Zandecki C, Ayana R, Pepermans E, Boonen K, et al. A short dasatinib and quercetin treatment is sufficient to reinstate potent adult neuroregenesis in the aged killifish. NPJ Regen Med. 2023;8(1):31. CrossRef
67. Hickson LTJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–56. CrossRef
68. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–63. CrossRef
69. Barinda AJ, Arozal W, Yuasa S. A review of pathobiological mechanisms and potential application of medicinal plants for vascular aging: focus on endothelial cell senescence. Med J Indones. 2022;31(2):132–40. CrossRef
70. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28(8):1556–68. CrossRef
71. Kang C. Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol Cells. 2019;42(12):821–7. CrossRef
72. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–78. CrossRef
73. López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. 2023;35(1):12–35. CrossRef
74. Wilson DM, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186(4):693–714. CrossRef
75. Evangelou K, Vasileiou PVS, Papaspyropoulos A, Hazapis O, Petty R, Demaria M, et al. Cellular senescence and cardiovascular diseases: moving to the “heart” of the problem. Physiol Rev. 2023;103(1):609–47. CrossRef