
INTRODUCTION
Progesterone (pregn-4-ene-3,20-dione), the main naturally occurring human progestogen, is synthesized mainly by the ovary and placenta, although it can also be synthesized by the liver and adrenal glands [1]. Progestogens have physiological and pharmacological effects on the neuroendocrine system, the reproductive system, the mammary glands, and the central nervous system, and among others [2]. According to the biopharmaceutics classification system, it is a class II drug, i.e., low solubility and high permeability [3].
Traditionally, it was accepted that progesterone had very little pharmacological activity by oral route, because when administered by this route, it undergoes high degradation because of the first-pass hepatic metabolism [4]. For this reason, progesterone was usually administered as an oily solution for intramuscular use, as a vaginal gel, as pessaries, or as suppositories, routes that were poorly accepted by patients and, therefore, did not guarantee compliance with the required therapy [5]. However, since the 80s, several reports appeared showing that oral absorption of progesterone improved with a decrease in particle size and variation in the composition of the dosage form [4,6–8]. An oral formulation appeared in the early 90s with adequate bioavailability [4], which proved to be effective mainly for the treatment of premenstrual syndrome [9]. The formulation contained micronized progesterone suspended in an oily base formed by a mixture of peanut oil and soy lecithin. The final product was a soft gelatin capsule, which was available in concentrations of 100 and 200 mg and is currently marketed with similar compositions.
The absorption of progesterone is mainly affected by its rate of dissolution; therefore, if it is possible to have a method that can detect and quantify significant differences between products with different particle sizes, it would become a fundamental tool for guide the development of similar formulations and use it routinely for quality control of manufactured batches. In addition, as part of the product development, it could also be used for the establishment in vitro–in vivo correlations, which is expected for active pharmaceutical ingredients class II in which the absorption is mainly affected by the dissolution rate of the active pharmaceutical ingredient from the drug; in the future, this would be able to make changes in the design or in the manufacturing process without requiring new in vivo studies [10].
Most dissolution tests in the United States pharmacopoeia (USP) for immediate or modified-release tablets and capsules use apparatus 1 or 2 (baskets or pallets, respectively). However, considering that the product being studied in this research are soft capsules containing progesterone suspended in an oily matrix, several problems may arise in the development of a dissolving method for this drug using the aforementioned apparatuses; these problems include: (a) the oily phase forming a separate layer on top of the dissolving media or (b) the appearance of oil droplets suspended in the media. All of this may adversely affect the release of the active pharmaceutical ingredient and may cause problems with the sampling and quantification of dissolved quantities, especially at the start of the dissolution test [11,12].
Based on the previous considerations, the aim of this work was to develop a dissolution method applicable to soft gelatin capsules containing 100 and 200 mg of progesterone in an oily base, which would be useful for both product development and quality control tests and would not exceed a 60 minutes duration.
MATERIALS AND METHODS
Materials
Progesterone USP standard; sodium lauryl sulfate (SLS, purity >99.0% in mass fraction) purchased from Sigma-Aldrich (St. Louis, MO); polysorbate 80 (Tween 80, Polyoxyethylene sorbitan monooleate), purchased from Croda (Parsippany, NJ); sodium acetate (purity between 99% and 101% in dry basis, mass fraction) provided by VWR chemicals (Radnor, PA); phosphoric acid (purity >85% mass fraction); sodium phosphate monobasic dihydrate (purity >99.5% in dry basis, mass fraction); sodium phosphate dibasic anhydrous (purity >99.95% in mass fraction); sodium acetate, anhydrous (purity >99% in mass fraction); sodium tetraborate decahydrate (purity >99.5% in mass fraction); anhydrous sodium hydroxide (purity >98% in mass fraction); hydrochloric acid (HCl, purity, 37% w/v) purchased from Merck (Darmstadt, Germany); glacial acetic acid (purity >99.5%) was supplied by Fluka (Buchs, Germany). The water used for all tests was type I. Two formulations of progesterone 100 mg [L PR190910, d (0.9) = 30 μm and lot PR110903 d (0.9) = 12 μm] were used, with two particle sizes, and as reference, the commercial formulation Prometrium® (Rottafarm S.r.l, Rome, Italy) 100 mg [L1PXKC06, d (0.9) = 28 μm].
Analytical methodology
High performance liquid chromatography (HPLC) equipment with diode array detector was used (Agilent 1200, USA). An injection volume of 10 μl of a 0.05 mg/ml progesterone solution, filtered by 0.45 μ filter (PVDF Whatman), was established in a Zorbax XDB C18 column (150 × 4.6 mm, 5 μm), with an oven temperature of 40°C. A methanol: water mixture (80:20), filtered at a flow of 1.0 ml/minute, was used as a mobile phase. The sample run time was 9 minutes, and detection was performed at a wavelength of 241 nm. Data were collected and analyzed by Agilent OpenLab A.02.02 software.
The analytical methodology was validated in terms of selectivity, linearity, accuracy, precision (repeatability and intermediate precision), stability of the analytical sample, and robustness, in accordance with USP recommendations [13].
Determination of sink conditions
Steady-state solubility for progesterone was determined in various aqueous media (water, simulated gastric juice, hydrochloric acid 0.1 N, acetates buffer pH 4.5, and phosphate buffer pH 6.8) and with increasing amounts of SLS (0.5%, 1.0%, 1.5%, 2.0%, 2.5%, 3.0%, 3.5%, and 4% w/v) and polysorbate 80 (0.1%, 0.3%, 0.5%, 1.0%, 1.5%, and 2% w/v).
In each media, an excess amount of progesterone was added (in duplicate) and stirred for 8 hours with a mechanical agitator (Vibromatic Selecta®, Spain); then, it was placed in the containers (vials of 20 ml) in a thermostated bath (Julabo, model SW23, Germany) at 37°C ± 1°C for 12 hours (previous trials showed that after 8 hours, we reached the balance). At the end, in all cases, an undissolved and a saturated solution of progesterone was found.
Samples were taken from the supernatant of each of the supersaturated solutions, filtered through 0.45 μm PVDF filters, and corresponding dilutions were made when required with each of the media and injected into the chromatograph. The samples were analyzed with the validated HPLC methodology, and the amount dissolved in the initial dilution, expressed as mg of progesterone per ml of each media, was determined.
For the calculation of sink conditions, two volumes were assumed: 250 ml, which is the maximum volume of dissolution media for USP apparatus 3 and 900 ml, which is the maximum volume for apparatus 1 and 2. To establish if sink conditions existed, the solubility found was divided by the maximum dose unit of progesterone capsules (200 mg) and multiplied by the volume of media, according to the dissolution apparatus used (converting the units to mg). The factor obtained is the number of times that the maximum unit dose is dissolved in the volume of media evaluated. It is generally accepted that there are sink conditions when the factor is three or greater [14,15]; i.e., at least three capsules of 200 mg can be dissolved in the volume of selected media.
Selection of the dissolution apparatus
The percentage of six capsules of progesterone 200 mg dissolved in USP dissolution apparatus 1, 2, and 3 was evaluated to that purpose, using the same phosphate buffer pH 6.8 solution media with 4% (w/v) of SLS in all three cases. In apparatus 1 (baskets) (Distek, model 6100, USA), a media of 900 ml was used at 150 rpm; in apparatus 2 (pallets) (Distek, model 6100, USA), with a volume of 900 ml at 100 rpm; and in apparatus 3 (oscillating cylinder) (Varian, model BIO-DIS, USA), in 250 ml, at 40 deeps per minute. Each of these apparatuses were used at the maximum recommended speeds according to USP [13].
Sinkers were used in all cases (QLA Lab. Accessories under part number: BSK008-JP) to prevent capsule floating during testing. The combination of solvent and surfactant was used as the solution media, which showed the highest equilibrium solubility for progesterone. A percentage of at least 85% of the active pharmaceutical ingredient dissolved was sought in a time of no more than 60 minutes.
Selection of dissolution conditions
Once the dissolution equipment was selected, a Plackett–Burman experimental statistical design (ESD) was used, with which eight experiments were performed with seven factors, each at a maximum level (+) and a minimum level (−). To define the test conditions, a screening of the variables that could affect the discriminatory power of the dissolution method was performed. In order to do this, two batches of the progesterone 200 mg product with differences in their particle size distribution were selected and evaluated under the established dissolution conditions according to the proposed ESD (Table 1). Six units from each batch were used to perform comparative dissolution profiles. The tests were carried out for 90 minutes, with a sampling interval of 15 minutes, taking into account the conditions presented in Table 2 and, as a response variable, the difference between the similarity factors (f2) obtained for each profile of each product [16], in each of the dissolution conditions according to the design was calculated.
The similarity factor (f2) was calculated using the following equation:
f2 = 50 × Log {[1+ (1/n) ∑t = 1n (Rt–Tt)2 ]−0.5 × 100}
where n, is the number of sampling points, Rt is the average percentage of dissolution of one of the products (the one used as a reference) at time “t.” Tt, is the average percentage of dissolution of the other product (of which you want to compare against the reference) at time “t.”
With the results of each factor evaluated in the ESD, the statistical analysis was performed, and the value of the effect was determined. The significance of each was estimated by the t-student test (α = 0.05) [17]. The experiments were carried out in random order, and in all the trials, between 3 and 5 drops of liquid silicone were added to each glass in order to avoid excessive foaming.
Assessment of the method’s discriminatory power
For this phase of the study, the aim was to evaluate how much the dissolution method differentiated between the product profiles that were considered different. For this purpose, the dissolution profiles of two test products were evaluated, for which it was determined that one met the bioequivalence criteria and the other did not meet the reference product. These test products corresponded to formulations that had their particle size modified. Product test 1: progesterone 100 mg batch PR190910, d (0.9) = 30 μm (bioequivalent to the reference product) and Product test 2: progesterone 100 mg batch PR110903 d (0.9) = 12 μm (it was not bioequivalent to the reference product), versus a reference batch Prometrium® 100 mg Batch 1PXKC06, d (0.9) = 28 μm. In product development studies, it had been defined that one of the most influential factors in bioavailability was the particle size of the active pharmaceutical ingredient in the suspension included in the capsules [8].
In both cases, the previously defined conditions were used to carry out the dissolution test, and it was expected for these profiles to yield an f2 value lower than 50, if they were actually different. From each of the batches, 12 units were analyzed, and the sampling intervals were selected according to preliminary tests carried out with each one of them (5, 15, 30, 45, and 60 minutes), ensuring that in all cases, at least 85% of the labeled substances was dissolved.
In order to have more information to analyze the results, the viscosity of the progesterone suspensions was evaluated, as some formulations were measured by varying their particle size (5, 18, 32, and 37 μm). The suspensions’ viscosity was carried out using a Brookfield DVIII equipment, with an accessory for handling small quantities of samples. The determination was made in triplicate at a temperature of 25°C.
RESULTS AND DISCUSSION
Determination of sink conditions
The results of the solubility study (data not shown), implicit in the sink condition-related factor, can be seen in Figure 1. Only those evaluated in the media with the SLS surfactant were included, since the experiments carried out with polysorbate 80, did not yield solubility values to provide sink conditions in the evaluated range.
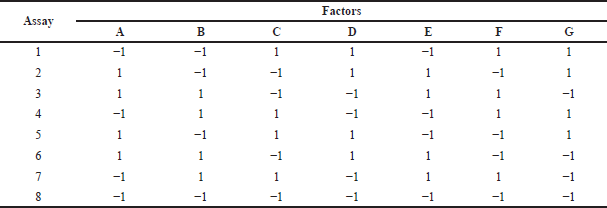 | Table 1. Matrix of the ESD used for the selection of dissolution conditions in apparatus 3. [Click here to view] |
Figure 1 shows that the solubility of progesterone in water is very low, and it does not vary with changes in pH between 1.2 and 6.8; this is expected considering that this drug is highly hydrophobic and does not present easily ionizable groups [14,18,19]. There was a significant increase in the concentration of progesterone in solution when each of the surfactants was used, especially in the case of SLS. Sink conditions (factor greater than or equal to 3) are guaranteed with a concentration of at least 1.5% SLS, in each of the media, assuming a dissolution volume of 250 ml or greater. If a 900 ml media volume is considered, sink conditions are reached ranging from a minimum of 0.5% SLS. In both cases, the highest calculated factor associated with sink conditions is obtained at a concentration of 4% SLS.
The importance of ensuring sink conditions is that the rate of dissolution of the drug, released from the dosage form, depends only on the behavior of the drug. This process is based on the diffusion phenomena, and therefore, it is driven by the concentration gradient. Therefore, if the media is saturated with the drug, there would be no gradient and, therefore, the process would be slowed down [20,21].
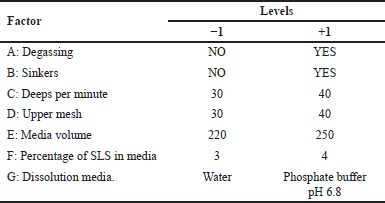 | Table 2. Factors selected for the ESD. [Click here to view] |
According to the results listed above, the value of 4% SLS is chosen, as the concentration at which the highest value of sink-factor was found, for the media volumes related to the three apparatuses. In addition, as mentioned in the methodology, the highest stirring conditions are chosen for each of the dissolution equipment available. As no differences were observed regarding the pH of the solution media, at this point, it was decided to continue with phosphate buffer pH 6.8 because the aim was to select a media with a similar pH to the progesterone absorption site, which occurs mainly at the duodenum [5,8].
Selection of the dissolution apparatus
Figure 2 shows the dissolution profiles for apparatus 1, 2, and 3, obtained under the previously chosen conditions. The results showed a significant difference for the same sample when apparatus 1 or 2 were used; the difference was more noticeable between the results of apparatuses 1 and 2, compared to apparatus 3.
For apparatuses 1 or 2, under the most drastic operation conditions of these devices, and even reaching the maximum percentage of SLS assessed (4%), the time required to reach at least 85% of the drug to be dissolved was longer than 2 hours and considering they are looking for a method of not more than 60 minutes (which could also be helpful for a routine quality control assay), the use of these devices was ruled out. In addition to the time required for dissolution, it was observed that the gelatin film dissolved in less than 30 minutes, but at the bottom of the glass, the oil content remained. It was dissolved in a very slow manner and it was found that at no time, it was properly dissolved to be fully incorporated into the media (this may explain the low dissolution rate observed). The time points found in apparatuses 1 and 2 coincide with those reported for a dissolution method for the same product, but in which cyclodextrins were used to dissolve it [22]. Although the effect of cyclodextrins cannot be compared to the SLS effect, it should be noted that perhaps the same mode of dissolution (due to the low incorporation of the sample in the media) may be the cause of such high time points in both conditions.
In apparatus 3, the percentage dissolved was almost 100% at 15 minutes time point. In this case, it was observed that the movement up and down the cell, entering and exiting the dissolution vessel, helped to disintegrate the oil content, which helped the incorporation of the solute into the media, and therefore, it facilitated its dissolution, in a media that was previously known to dissolve up to 10 times the 200 mg, corresponding to the dose of progesterone contained in the capsule.
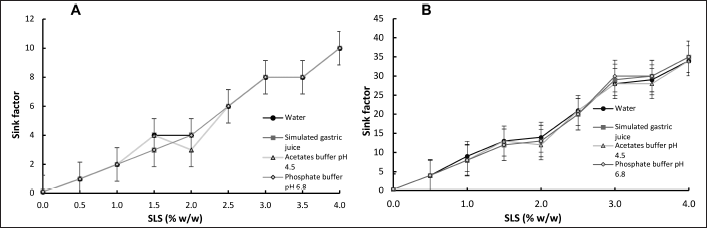 | Figure 1. Relationship between SLS concentration and sink conditions in various dissolution media, at 37°C ± 1°C for 12 hours. A. Volume of dissolution media used: 250 ml; B. Volume of dissolution media used: 900 ml. SLS: sodium lauryl sulfate; each value corresponds to average ± SD; n = 2. [Click here to view] |
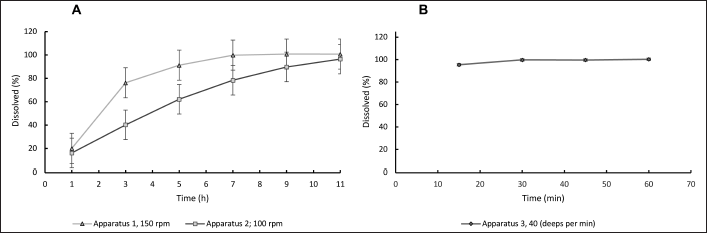 | Figure 2. Progesterone dissolution profile 200 mg. A. Apparatus 1 (150 rpm) and Apparatus 2 (100 rpm), using 900 ml phosphate buffer pH 6.8 with 4% SLS; B. Apparatus 3, 40 deeps per minute, using 250 ml phosphate buffer pH 6.8 with 4% SLS. SLS: sodium lauryl sulfate; each value corresponds to average ± RSD; n = 6. [Click here to view] |
Based on these results, apparatus 3 was selected to carry out the following stages of the study.
Selection of dissolution conditions
For the selection of dissolution conditions, a Plackett-Burman design of eight experiments was used to assess the effects of the various factors related to apparatus 3 on dissolution speed, according to what was stated in the methodology.
According to preliminary assay results, 90 minutes duration and 15 minutes sampling were defined to achieve at least 85% of the pharmaceutical active ingredient to be dissolved. As explained in the methodology, the experiments were performed for the two progesterone products of 200 mg, which have been manufactured with differences in their particle size. One of the products had a particle size distribution in which 90% of its particles were smaller than 120 μm, while in the second product, 90% of its particles were smaller than 250 μm. The first of these was used as a reference product for the calculation of f2. With the data obtained, a bar chart was constructed to show the statistical significance and the sign of each factor on the selected response variable.
Figure 3 shows that variables such as degassing (Factor A) or deeps per minute (Factor C) in the assessed ranges did not have a statistically significant effect on the response, that is, they did not significantly influence the discriminatory power of the dissolution method. However, it is always recommended to degas the media since it is a good practice to ensure reproducibility of results between laboratories, and in the case of deeps per minute, a value of 30 deeps per minute is recommended to reduce the possibility of foaming.
On the other hand, the following factors had a statistically significant effect on the response variable: sinkers (B), upper mesh (D), media volume (E), percentage of SLS in media (F), and dissolution media (G). The presence of sinkers increased the percentage of difference found between two different products; it is believed that this is due to the fact that the sinker ensures the capsule is always in the bottom of the glass during the test, leading to a decreased variability of the data and was able to ensure that the characteristics of the solution obtained mostly depended on the performance of the dosage form. Therefore, it was decided to include the use of the sinker in the dissolution method.
It was observed that increasing the number of the upper mesh, from 30 to 40, had a negative effect on the discriminatory power of the method. This observation coincides with the USP recommendation to use an upper mesh of a smaller number than the one used in the lower mesh; thereby, aggregates and dissolved material inside the cell could leave through the upper part and, therefore, to be incorporated into the dissolution media. It was decided to use a lower mesh of 40 and an upper mesh of 30.
Regarding the percentage of SLS, although it was found that there was a statistically significant negative effect on the response to an increase in the concentration of SLS, in order to maintain a short time-lapse for the test, it was decided to continue working with 4%. During the assessment phase of the discriminatory power of the method, it was observed that it was necessary to decrease this percentage, it was considered to do so.
Finally, regarding the dissolution media, it was set to be 250 ml of phosphate buffer at pH 6.8. Although these two parameters (media volume and pH) had a significant negative effect on the response, using a volume of 220 ml, it was observed that the cell was not fully immersed in the vessel at the time it went down during the deep and the phosphate buffer pH 6.8, as mentioned above, is the pH of the media to facilitate the absorption of progesterone.
With these selected conditions, a test time of 45 minutes was established, in which at least 85% of the progesterone was guaranteed to be dissolved from soft gelatin capsules with 200 mg of the active pharmaceutical ingredient.
Assessment of the method’s discriminatory power
This assessment was carried out using the dissolution method under the conditions established above, using the products mentioned in the methodology: one batch that showed to be bioequivalent and another that failed this study, compared to the reference product. The results are shown in Figure 4.
The results of the dissolution profiles (Fig. 4) showed the same trend as the bioequivalence data (data not shown). In all the profiles obtained, for both test and reference products, coefficient of variation (CV%) values are high (greater than 20%) in the 5 and 15 minutes samples. The reason for these values of CV%, is that before the 10 to 12 minutes time points, the soft gel disintegration is not complete and is particularly variable; this is a common trend for the soft capsules in comparison to a tablet or immediate-release hard capsule, where after about 6 to 8 minutes, they have broken down in its entirety and the dissolution rate at early times (unless they are sampled before 5 minutes, approximately), is not significantly affected by the effect of material trapped by the covering film or external coating.
In the reference profile and test 1 profile (which were bioequivalent), there is practically an overlap of the two curves, which is verified with the result of f2 between these two profiles that is greater than 50 (f2 = 64). On the other hand, the dissolution profiles of test 2 versus the reference presented a difference in the amount of active ingredient dissolved in the first time points assessed, although after 45 minutes it is practically dissolved reaching 100%, but in general terms, the profiles are different, that is, f2 is lower than 50 (f2 = 30).
Something that is striking is a decrease in the dissolution rate when the particle size is reduced; this is contradictory to what would be expected in general for low-solubility active pharmaceutical ingredients [23] and even to what is reported in the literature for progesterone [8]. In general, when it comes to low-solubility active pharmaceutical ingredients such as progesterone, the decrease in particle size should increase the dissolution rate, and therefore, the biopharmaceutical parameters, such as area under the curve and maximum concentration (Cmax). In order to try to understand the reason for this behavior in the product, test 1 and test 2 solutions were assessed again to observe the behavior during the assay.
From the results of this new assay, it was observed that for both batches, the gelatin disintegrated within 15 minutes, but for test 2, the dispersion of the oily suspension in the dissolving media was much more difficult to achieve than for test 1 or the reference material. This behavior appeared to be related to the particle size of the drug. Apparently, the finer particles of the active pharmaceutical ingredient for batch test 2, adsorb much more of the oil phase of the suspension, and therefore, it creates much more viscous suspensions than those achieved with product test 1 and reference product, which had progesterone with larger particle size. To verify this, we measured the viscosity of the suspensions of both products test 1 (13,587 cps ± 3.0%), product test 2 (38,347 cps ± 1.8%), and the reference product (16,100 cps ± 4.3%); in addition, they were compared regarding the viscosity obtained for formulations with the same composition, but with different particle sizes (Fig. 5).
According to the results, it was observed that, in the assessed range, the viscosity of the suspension increases as the particle size decreases, and this increase in viscosity may explain why the dissolution of the product decreases accordingly.
It is believed that the previous findings do not contradict what has been reported in the literature regarding the fact that micronization of progesterone increases its bioavailability [8]. As expected for a low-solubility drug, it is suggested that decreasing the particle size favors the bioavailability and the dissolution rate of progesterone in an appropriate particle size range. Excessive micronization can have the opposite effect when the active pharmaceutical ingredient is incorporated into a suspension, as these finer particles can adsorb more of the oily phase, creating a more viscous suspension, which would be more difficult to disperse in the aqueous media. This behavior would result in a decrease in the dissolution rate that would lead to a lower absorption rate and, therefore, lower values in the biopharmaceutical parameters.
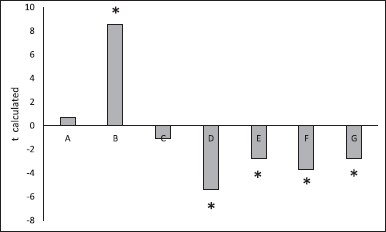 | Figure 3. Influence of variables related to the dissolution method (Apparatus 3), on the similarity factor (response variable). (A) Degassing; (B) Sinkers; (C) Deeps per minute; (D) Upper mesh; (E) Media volume; (F) Percentage of SLS in media; (G) Dissolution media (factors marked with an asterisk have a statistically significant effect α = 0.05). [Click here to view] |
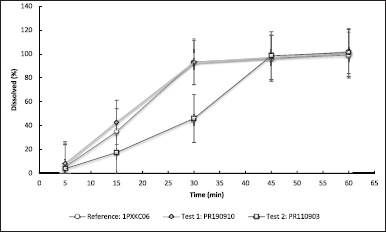 | Figure 4. Dissolution profile obtained for each of the batches evaluated, using the proposed dissolution methodology (USP apparatus 3, 30 deeps per minute, using 250 ml phosphate buffer pH 6.8 with 4% SLS at 37°C ± 1°C for 60 minutes, using sinkers, upper mesh No. 30 and lower mesh No 40). Each value corresponds to average ± RSD; n = 12; product test 1: progesterone 100 mg batch PR190910, d (0.9) = 30 μm (bioequivalent to the reference product), product test 2: progesterone 100 mg batch PR110903 d (0.9) = 12 μm (it was not bioequivalent to the reference product), reference batch Prometrium® 100 mg Batch 1PXKC06, d (0.9) = 28 μm. [Click here to view] |
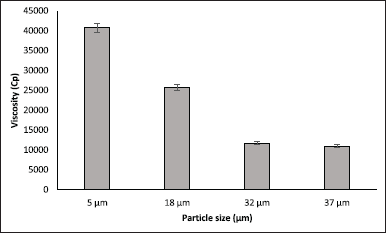 | Figure 5. Influence of progesterone particle size on suspension viscosity (At 25°C; spindle 6, 10 rpm). Each value corresponds to average ± RSD; n = 3. [Click here to view] |
The results obtained using the proposed dissolution method in samples of products with significant differences in particle size of the dispersed phase showed adequate discriminatory power to differentiate distinct batches and to establish similarity when applying the methodology to batches considered similar based on their in vivo comparison.
Based on the profiles obtained, the best time to establish a possible specification of the dissolution test for a quality control test is 30 minutes, since at that time, the dissolution percentages of the products that failed the bioequivalence test are significantly differentiated. Thus, bioequivalent batches such as reference and test 1 achieve a percentage of dissolved progesterone of approximately 90% for 12 units tested in each one of them, while the one that was not bioequivalent (i.e., test 2), only reaches approximately 40%, this being consistent with the biopharmaceutical parameters that were found during the in vivo comparison of these batches (results are not shown).
Once the specification time in 30 minutes was selected, the next step was to define a possible Q value at this time point, and to achieve this, it was necessary to observe the minimum individual values of each product. The capsule with the minimum dissolution value between the reference batch and test batch 1 was 87%, which allowed us to propose a Q value of 80%, so the specification would be as follows: Q = 80% for the 30 minutes time point. This means that six units analyzed in a batch, during the dissolution test as part of a quality control test, should yield dissolution percentages as established on the label, of at least 85% (Q + 5%) [13].
CONCLUSION
It was possible to define a dissolution method for the product development phase of progesterone soft gelatin capsules, using USP apparatus 3, with a volume of 250 ml media composed of phosphate buffer pH 6.8 and 4% SLS; upper mesh No. 30 and lower mesh No 40; operating at 30 deeps per minute; using sinker for each capsule; with a duration of 60 minutes to obtain a dissolution of 85% and with the addition of 3 to 5 drops of liquid simethicone in each dissolution glass to prevent foaming. The proposed dissolution method allowed to discriminate batches with differences in particle size that affect the bioavailability of progesterone. This method is also useful as a product quality control test and a (Q) of 80% is suggested at the 30 minutes time point as a possible specification.
ACKNOWLEDGMENTS
The authors would like to express gratitude to Procaps Laboratory for financially supporting this study. The sponsor was not involved in the study design, collection, analysis, and interpretation of information, writing of the manuscript, and the decision to submit the paper for publication.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Roberts LJ. Goodman & Gilmans. The pharmacological basis of therapheutics. 10th ed. New York, NY: McGraw-Hill; 2001.
2. Whitehead MI, Townsend PT, Gill DK, Collins WP, Campbell S. Absorption and metabolism of oral progesterone. Br Med J. 1980;280:825–7. CrossRef
3. Dahan A, Miller JM, Hoffman A, Amidon GE, Amidon GL. The solubility-permeability interplay in using cyclodextrins as pharmaceutical solubilizers: mechanistic modeling and application to progesterone. J Pharm Sci. 2010;99:2739–49. CrossRef
4. Pucci V, Bugamelli F, Mandrioli R, Luppi B, Raggi MA. Determination of progesterone in commercial formulations and in non-conventional micellar systems. J Pharm Biomed Anal. 2003;30:1549–59. CrossRef
5. Hargrove JT, Maxson WS, Wentz AC. Absorption of oral progesterone is influenced by vehicle and particle size. Am J Obstet Gynecol. 1989;161:948–51. CrossRef
6. Chakmakjian Z, Zachariah N. Bioavailability of progesterone with different modes of administration. J Reprod Med. 1987;32:443–8.
7. Cheema M, Palin KJ, Davis SS. Lipid vehicles for intestinal lymphatic drug absorption. J Pharm Pharmacol. 1987;39:55–6. CrossRef
8. Kincl FA, Ciaccio LA, Benagiano G. Increasing oral bioavailability of progesterone by formulation. J Steroid Biochem. 1978;9:83–4. CrossRef
9. Freeman EW, Rickels K, Sondheimer SJ, Polansky M. A double-blind trial of oral progesterone, alprazolam, and placebo in treatment of severe premenstrual syndrome. JAMA. 1995;274:51–7. CrossRef
10. ICH Q8 (R2). Harmonised tripartite guideline: pharmaceutical development: scientific guideline. Proceedings of the International Conference on Harmonization, Geneva, Switzerland; 2009. Available from: https://www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development
11. Damian F, Harati M, Pathak V, Schwartzenhauer J, Durham D, Quiquero V, et al. Development of a discriminating dissolution method for immediate-release soft gelatin capsules containing a BCS class II compound. Dissolution Technol. 2016;23:6–13. CrossRef
12. Hu J, Kyad A, Ku V, Zhou P. A comparison of dissolution testing on lipid soft gelatin capsules using USP apparatus 2 and apparatus 4. Dissolution Technol. 2005;12(2):6–9. CrossRef
13. USP. United States pharmacopeia and national formulary USP 42- NF 37. Rockville, MD: The United States Pharmacopeial Convention; 2019.
14. Martin GP, Gray VA. Selection of dissolution medium for QC testing of drug products. J GXP Compliance. 2011;15:29–35.
15. Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms. Proceedings of the Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Washington, DC; 1997. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/dissolution-testing-immediate-release-solid-oral-dosage-forms
16. Moore JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Technol. 1996;20:64–75.
17. Connors K, Amidon G, Stella V. Chemical stability of pharmaceuticals: a handbook for pharmacists. 2nd edition. New York, NY: John Wiley & Sons; 1986.
18. Galia E, Nicolaides E, Hörter D, Löbenberg R, Reppas C, Dressman JB. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 1998;15:698–705. CrossRef
19. Rohrs B. Dissolution method development for poorly soluble compounds. Dissolution Technol. 2001;8(3):6–12. CrossRef
20. Siepmann J, Siepmann F. Sink conditions do not guarantee the absence of saturation effects. Int J Pharm. 2020;577:119009. CrossRef
21. Sinko PJ. Martin’s physical pharmacy and pharmaceutical science. In: Troy D, editor. Physical chemical and biopharmaceutical principles in the pharmaceutical sciences. 6th edition. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
22. Besins A, Besse J, inventors, Besins Healthcare Luxembourg SARL, assignee. Pharmaceutical composition based on micronized progesterone, preparation method and uses thereof. United States Patent US20110135719A1. 2012.
23. Singh S, Singh N, Allawadi D, Arora S. Techniques for bioavailability enhancement of BCS class II drugs: a review. Int J Pharm Chem Sci. 2013;2:1092–101.