
INTRODUCTION
Dopamine (DA) has a catechol ring. Dopaminergic neural system dysfunction is associated with various CNS disorders, such as paranoia, schizophrenia, autism, and Parkinson’s disease [1,2]. In the evaluation of the effectiveness of drugs designed to treat and control DA-related disorders, determining changes in the baseline level of DA in the brain is significant. Therefore, DA in the brain needs to be determined [3]. For quantitative DA estimation, various analytical methods, including GC-ECD [4,5], high-performance liquid chromatography (HPLC) [6–9], LC-mass spectrometer (MS)/MS [10–12], and fluorimetry [13] have been published. Mustafa et al. [12] reported a validated LC-MS/MS system for the study of DA in the rat brain with a limit of detection (LOD) of 0.278 ng ml−1 and a limit of quantification (LOQ) of 0.844 ng ml−1. These investigations have also further stressed the need for a fast, sensitive, and selective method for detecting DA with shorter retention times and lower quantification limits. Moreover, the analysis carried out by Mustafa et al. [12] was done on ultra-HPLC (UHPLC)/electrospray ionization (ESI)-Q-TOF-MS system, whereas in the present study, UHPLC-Xevo-triple quadrupole (TQD)-MS/MS system has been used.
Indeed, detection with Xevo TQD reduces the unpredictability and simplifies the bioanalytical procedure. RADARTM technology, in the detection system, allows for better matrix interference assessment by assembling the data simultaneously in full scan and multiple reaction monitoring (MRM) modes. Also, all the detectable ions, including parent ions, can automatically detect in both positive and negative MS scans, which consequently provides analysts with a deep understanding of the analyte under detection, which cannot be achieved with the traditional bioanalytical approach. Also, Xevo TQD collects and interprets the data quickly because of in-built T-WaveTM collision cell technology, making it truly compatible with the narrowest of response peak widths [14].
The ultra-performance liquid chromatography (UPLC)-Xevo-TQD-MS/MS method has also been reported to quantify certain drugs like apatinib [15], N-methylcytisine [16], ledipasvir [17], cabozantinib [18], etc.
EXPERIMENTAL SECTION
Materials and reagents
DA and adrenaline (Adr) were purchased from Sigma Aldrich (Seelze, Germany). MS grade formic acid (FA) (purity >98%), ammonium acetate, and acetonitrile (ACN) were procured from Fluka Analytical (Sigma-Aldrich, The Netherlands). Milli-Q system (Millipore Corp. USA) was utilized to purify the deionized water.
UHPLC conditions
UHPLC separation was carried out using an ACQUITY UPLC system (Waters Corp. USA), coupled with a dual solvent delivery system and ACQUITY UPLC Ethylene Bridged Hybrid C18 column (50.0 × 2.1 mm; 1.7 μm). The mobile phase consisted of ACN and FA (0.1% w/v) [30: 70; v/v], with isocratic elution at a constant flow rate of 0.3 ml minute−1. The total chromatographic run time was 3.0 minutes, and 5 μl of sample solution was injected in each run.
Xevo-TQD-MS conditions
A Xevo-TQD MS outfitted with an ESI source (Waters Corp.) in positive mode was employed for the quantitative analysis of DA, Adr, and internal standard (IS) in rat brain homogenate. Bioanalysis was performed using the MRM transitions at 154.084 m/z →91.097 m/z for DA and 184.104 m/z→107.089 m/z for IS (Fig. 1), with MS and MS/MS mode collision energy set at 3 and 30 V, respectively. For ion monitoring, the source temperature was maintained at 130°C, with capillary voltage at 3.50 kV and 1,000 l/hour flow rate for desolvation gas. Data collection and instrument management were performed on MassLynx V 4.1 SCN918 software (Waters Corp.).
Preparation of standards
For calibrations and quality control (QC) standards, a 1 mg ml−1 stock solution of DA was prepared using methanol, and further diluted with the solvent mixture, containing ACN and water (50:50, v/v), to prepare working solutions at different concentration levels. Similarly, QC samples and calibration standards for rat brain homogenate were obtained by diluting the blank rat brain homogenate with corresponding working solutions. The calibration curve was prepared using 0.577, 1, 5, 10, 50, 125, 250, 600, and 800 ng ml−1 as final concentrations of DA in rat brain homogenate. The concentrations of QC samples in brain homogenate were 600, 125, 5, and 1 ng ml−1 for high-quality control (HQC), middle-quality control (MQC), low-quality control (LQC), and lower limit of quantification control (LLOQC), respectively. Likewise, DA, 1 mg ml−1 stock solution of IS (Adr) was further diluted to 4 μg ml−1 using ACN:water (50:50 v/v) to obtain the IS working solution. All prepared solutions were refrigerated at 2°C–8°C for further use.
Brain homogenate sample preparation
A 200 μl aliquot of brain homogenate after thawing at 25°C, was taken in a 1.5 ml Eppendorf tube with 50 μl of IS working solution (4 μg ml−1). The protein precipitation technique was carried out to extract the DA from a biological sample using 0.5 ml of 5%w/v FA. After vortexing for 2 minutes, 10 μl of the mixture was extracted using ethyl acetate (1 ml). Upon evaporation, the sample was reconstituted with 50 μl of ACN and FA (30: 70, v/v). The 5 μl of the resultant mixture was then injected into the UHPLC system for analysis using auto-sampler vials.
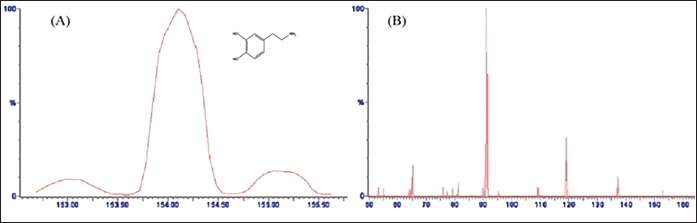 | Figure 1. Optimized mass spectrum of (A) DA precursor ion at cone voltage 24 V (protonated precursor [M + H]+ ion at m/z 154.084) with chemical structure, and (B) optimized product ion spectrum of DA at collision energy 22 V (major fragmented product ion at m/z 91.097) showing fragmentation transitions. [Click here to view] |
Bioanalytical method validation
Prior to determining DA in brain homogenate samples, the developed UHPLC method was exclusively validated for its linearity, stability, sensitivity, accuracy, precision, matrix effect, and recovery. The calibration plots were constructed to evaluate the linearity using the peak area ratio of DA to IS versus the analyte concentration via the least squares linear regression technique. The selectivity of the method was assayed by analyzing six lots of blank biological samples, alone and spiked with DA and IS. The LLOQ, defined as the lowest concentration of the standard curve, was determined using the equation mentioned below. The accuracy of the method as recovery was examined by analyzing the DA standard solution (5 ng ml−1) at concentration levels of 50% and 100% (n = 3). The precision was assayed by estimating all four QC concentrations. Both inter- and intra-day precision and accuracy were evaluated on the same day and three different days, respectively, in six replicates. The accuracy was calculated as a percentage of deviation of the measured mean drug concentration to the theoretical concentration. The method’s robustness was assayed by applying minor variations in UHPLC conditions using all three QC concentrations. However, for ruggedness assessment, different columns (but of the same type) were employed using a single batch of precision by another analyst on the same instrument in six replicates [19].
The recovery of DA was determined by analyzing the mean area response of samples spiked before extraction to that of biological samples spiked after extraction, in replication of six. In the same way, the recovery of IS was determined [15]. The matrix effect was explored using the post-extraction spike method. The matrix effect was calculated as a ratio (A/B × 100), in which A was the peak area of the blank sample spiked with MQC and B was the equivalent peak area attained by the spiking standard in the mobile phase.
LOD and LOQ were determined using the following formula:
Ex vivo stability
The stability of DA in the biological sample was evaluated at the level of HQC and LLOQC, in replication of six, exposed to the diverse environment (storage period and temperature). The precisions were lower than 15%, and the accuracy of 85%–115% for both levels was adapted as stable.
Long-term stability
Biological samples spiked with standard DA having LLOQC and HQC levels, stored at −80°C for 35 days, were used to determine the long-term stability, using six sets each.
Freeze-thaw stability
After three consecutive freeze-thaw cycles from −20°C to + 25°C, six sets of HQC and LLOQC were evaluated for DA concentration in brain homogenate.
Bench-top stability
Under optimal conditions, six sets of LLOQC and HQC were kept at the bench top for 24 hours to analyze the bench-top stability at 25°C.
Post-processing stability
After 24 hours of exposure at 10°C in an autosampler, six replicates of LLOQC and HQC samples were processed and analyzed to assess the short-term stability [12].
In vivo study
Experimental animal
Adult Wistar rats (weighing 220−240 g) of either sex, obtained from the central animal house of Jamia Hamdard, New Delhi, were used in the experiment, after approval from the Institutional Animal Ethics Committee (IAEC). All animals were fed with rat food and water for 7 days and fasted overnight 1 day before experimentation.
Experimental protocol
Animals were divided into four groups (three rats each). Group I was taken as normal; Group II as control; Group III was treated with selegiline solution, administered intravenously (18 μg/day dose mixed in 500 μl of isotonic solution); and Group IV was treated with selegiline nanoemulsions, administered intranasally (dose equivalent to 18 μg/day). This nanoemulsion was prepared by the method explained by Kumar et al. [20]. Rats from different groups were euthanized, and their brains were isolated. The supernatants obtained from homogenizing the brain with centrifugation at 15,000 rpm for 20 minutes were then preserved at −80°C for analysis.
RESULTS AND DISCUSSION
UHPLC/ESI-Xevo-TQD-MS/MS analysis
The chromatographic settings were optimized to increase the DA signal intensity, improve peak shape, and reduce the run time. DA showed positive precursor ions [M + H]+ in ESI + mode due to the presence of basic nitrogen in the DA molecule. An optimum separation of analytes was carried out using ACN: FA (0.1% w/v) (30: 70; v/v) with isocratic elution at 0.3 ml minute−1 flow rate for an acceptable run time of 3 minutes. The MS spectra for DA displayed a peak of product ions at m/z 154.084 with a fragmented product ion peak at m/z 91.097 (Fig. 1A and B). However, IS showed [M + H]+ ion peak at m/z 184.104 with the most abundant product ions at m/z 107.089 (Fig. 2A and B). The obtained main product ion peaks were further used for the final quantification. The capillary voltage of 3.50 kV was applied to examine the precursor ions with 22 and 20 V as optimal collision energies for both DA and IS, respectively.
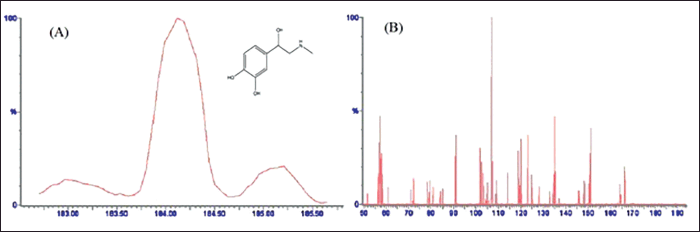 | Figure 2. Mass spectrum of (A) adrenaline (IS) precursor ion at cone voltage 20 V (protonated precursor [M + H]+ ion at m/z 184.104 with chemical structure, and (B) optimized product ion spectrum of IS at collision energy 20 V (major fragmented product ions at m/z 107.089) showing fragmentation transitions. [Click here to view] |
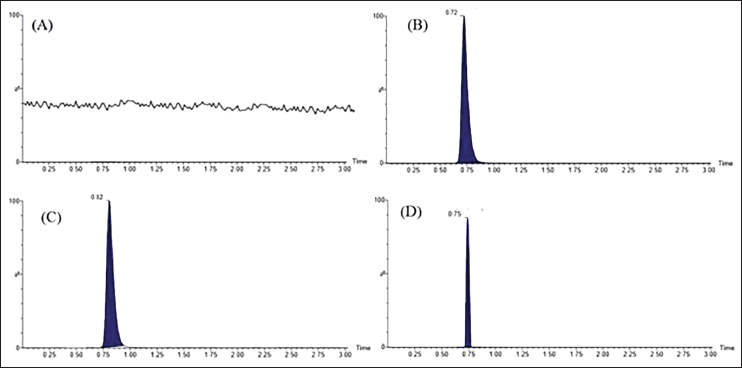 | Figure 3. Typical chromatograms of (A) blank brain homogenate, (B) DA (250 ng ml−1), and (C) IS (250 ng ml−1) extracted after spiking with Wistar rat-brain homogenate by selected reaction monitoring scan mode. In the same way, (D) showed extracted DA obtained after treatment with selegiline nanoemulsion (administered intranasally). The endogenous DA showed a slight shift in retention time. [Click here to view] |
To achieve maximum recovery, four solvents (ethyl acetate, chloroform, methanol, and dichloromethane) were tested. It was observed that ethyl acetate alone provided more recovery (>88%) for analytes. The chromatogram of the blank homogenate sample was displayed in Figure 3A. The DA (250 ng ml−1) eluted at 0.72 minutes (Fig. 3B), and IS (250 ng ml−1) eluted at 0.72 minutes (Fig. 3C), whereas the DA-spiked biological sample obtained from treatment group IV was retained at 0.75 minutes (Fig. 3D).
Bioanalytical method validation
The developed bioanalytical method was validated using various parameters as described in the ICH Q2 (R2) guidelines [21].
Selectivity
The isocratic mobile phase, ACN and FA (0.1% w/v) (30:70; v/v), effectively separated both DA and IS with no interfering endogenous peaks. Figure 3 shows the typical MRM chromatograms in SRM scan mode of blank, spiked samples, and biological samples demonstrating the selectivity of the method.
Linearity
A good linear regression between peak area ratios and DA concentrations in brain homogenate (ranging from 0.577 to 800 ng ml−1) was observed. The typical calibration curve equation was found to be Y = 0.001X – 0.005, where Y represents the peak area ratio and X represents the DA concentration in the biological sample.
Recovery, accuracy, and precision
The extraction recovery of the spiked DA biological sample was 85.41%, 83.70%, and 81.04% (Table 1), whereas the recovery of IS was found to be 90.07%. The accuracy of both intra- and inter-batches was within 92.60%–99.72%. Similarly, precision (%RSD) for both intra- and inter-batch ranged from 0.43 to 2.29 for all the QC levels (Table 2). The obtained values meet the criteria for determining biological samples according to USFDA guidance, indicating the method is accurate and precise.
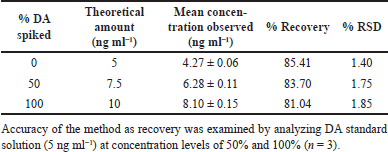 | Table 1. Recovery data for DA. [Click here to view] |
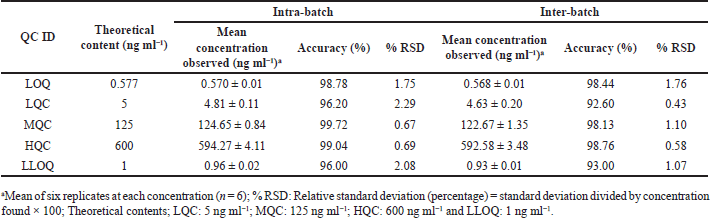 | Table 2. Precision and accuracy data. [Click here to view] |
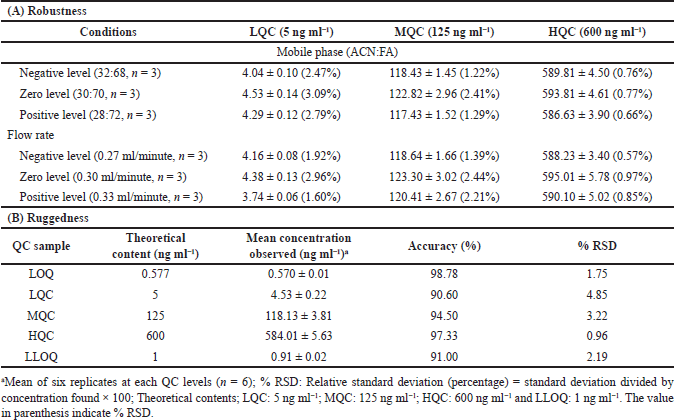 | Table 3. Robustness and ruggedness of the method. [Click here to view] |
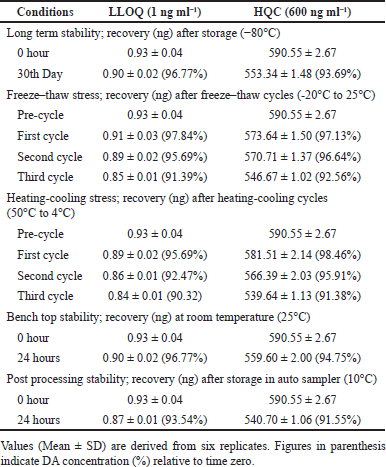 | Table 4. Ex vivo stability data. [Click here to view] |
 | Table 5. Estimation of DA in brain (ng ml−1). [Click here to view] |
Robustness
The correctness of the developed method was confirmed by evaluating the influence of diverse constraints on the %RSD and recovery of DA, including different flow rates (0.27, 0.30, and 0.33 ml minute−1) and variation in mobile phase concentrations (ACN:FA v/v, 32:68, 30:70, and 28:72). Small values of %RSD (0.57%–3.09%) were observed (Table 3) after diminutive but premeditated modification in the developed method specifying the method’s robustness.
Ruggedness
Different analysts evaluated the accuracy and precision of a batch by employing diverse columns. The average accuracy for DA ranged between 90.60 and 98.78 and %RSD from 0.96 to 4.85, as shown in Table 3.
Matrix effect
At LQC, 93.02% (n = 6; %RSD 4.66), the matrix effect of DA was observed, but at HQC, it was 96.11% (n = 6; %RSD 4.72). The value of %RSD was less than 5%, indicating that the method has no matrix effect.
LOD and LOQ
Using the equation, both LOD and LOQ were found to be 0.190 and 0.577 ng ml−1, respectively.
Ex vivo stability
The results of all stability studies of DA in brain homogenate samples at LLQC and HQC were displayed in Table 4. After 35 days, 96.77% (LLOQC) and 93.69% (HQC) of DA were recovered. After 3 freeze-thaw cycles, the recovery was in the range of 91.39%–97.84% (LLOQC) and 92.56%–97.13% (HQC). In benchtop stability assay, 96.77% and 94.75% of DA were recovered in LLOQC and HQC, respectively. However, in the post-processing stability assay, the DA recovery was 93.54% (LLOQC) and 91.55% (HQC). It was found that under all storage conditions, DA was stable.
In vivo study
A fast and efficient UHPLC Xevo TQD MS/MS technique was developed and validated successfully to analyze DA in rat brain tissue (Table 5). The average DA concentration in the normal group was 20.49 ± 4.15 (ng ml−1). In the control group rats, the DA level was found to be 8.59 ± 1.00 ng ml−1, whereas the selegiline solution-treated group showed 12.07 ± 1.98 ng ml−1. Selegiline nanoemulsion (administered intranasally) treated rats showed significantly (p < 0.01) increased DA level (16.61 ± 3.06 ng ml−1). The results suggested that selegiline nanoemulsion administered intranasally upregulates the level of DA in rat brain.
CONCLUSION
The Xevo-TQD –MS-MS method has been developed for the routine evaluation of DA in rat brain. The method has efficient separation and a short analysis time. Our method has a low retention time, thus leading to less consumption of the mobile phase. The method discussed is unique, sensitive, adequately precise, and accurate.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
ETHICAL APPROVAL
Animals used for the present experiment were approved by the IAEC of Jamia Hamdard, New Delhi, with approval number 1123.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Koller WC, Rueda MG. Mechanism of action of dopaminergic agents in Parkinson’s disease. Neurology. 1998;50(6 SUPPL.6):S11–14.
2. Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol Psychiatry. 2004;9(6):557–69.
3. Su F, Wang F, Zhu R, Li H. Determination of 5-hydroxytryptamine, norepinephrine, dopamine and their metabolites in rat brain tissue by LC-ESI-MS-MS. Chromatographia. 2009;69(3–4):207–13.
4. Ko H, Lahti RA, Duchamo DJ, Royer ME. A GC-MS procedure for the measurement of dopamine in mouse striatal tissue. Anal Lett. 1974;7(4):243–55.
5. Holdiness MR, Rosen MT, Justice JB, Neill DB. Gas chromatographic-mass spectrometric determination of dopamine in subregions of rat brain. J Chromatogr A. 1980;198(3):329–36.
6. Cannazza G, Di Stefano A, Mosciatti B, Braghiroli D, Baraldi M, Pinnen F, et al. Detection of levodopa, dopamine and its metabolites in rat striatum dialysates following peripheral administration of L-DOPA prodrugs by mean of HPLC-EC. J Pharm Biomed Anal. 2005;36(5):1079–84.
7. Karimi M, Carl JL, Loftin S, Perlmutter JS. Modified high-performance liquid chromatography with electrochemical detection method for plasma measurement of levodopa, 3-O-methyldopa, dopamine, carbidopa and 3,4-dihydroxyphenyl acetic acid. J Chromatogr B. 2006;836(1–2):120–3.
8. Muzzi C, Bertocci E, Terzuoli L, Porcelli B, Ciari I, Pagani R, et al. Simultaneous determination of serum concentrations of levodopa, dopamine, 3-O-methyldopa and α-methyldopa by HPLC. Biomed Pharmacother, 2008;62(4):253–8.
9. Najmanová V, Rambousek L, Syslová K, Bubeníková V, Šlamberová R, Valeš K, et al. LC-ESI-MS-MS method for monitoring dopamine, serotonin and their metabolites in brain tissue. Chromatographia. 2011;73(SUPPL. 1):143–9.
10. Carrera V, Sabater E, Vilanova E, Sogorb MA. A simple and rapid HPLC-MS method for the simultaneous determination of epinephrine, norepinephrine, dopamine and 5-hydroxytryptamine: application to the secretion of bovine chromaffin cell cultures. J Chromatogr B. 2007;847(2):88–94.
11. El-Beqqali A, Kussak A, Abdel-Rehim M. Determination of dopamine and serotonin in human urine samples utilizing microextraction online with liquid chromatography/ electrospray tandem mass spectrometry. J Sep Sci. 2007;30(3):421–4.
12. Mustafa G, Ahmad N, Baboota S, Ali J, Ahuja A. UHPLC/ESI-Q-TOF-MS method for the measurement of dopamine in rodent striatal tissue: a comparative effects of intranasal administration of ropinirole solution over nanoemulsion. Drug Test Anal. 2013;5(8):702–9.
13. Wang HY, Sun Y, Tang B. Study on fluorescence property of dopamine and determination of dopamine by fluorimetry. Talanta. 2002;57(5):899–907.
14. Vogliardi S, Tucci M, Nalesso A, Stocchero G, Ferrara SD, Favretto D. HPLC-HRMS vs UPLC-MS/MS for a systematic toxicological screening in hair. Toxicol Anal Clin. 2014;26(2):S15–6.
15. Zhou Y, Wang S, Ding T, Chen M, Wang L, Wu M, et al. Evaluation of the effect of apatinib (YN968D1) on cytochrome P450 enzymes with cocktail probe drugs in rats by UPLC-MS/MS. J Chromatogr B. 2014;973:68–75.
16. Wang S, Wu H, Huang X, Geng P, Wen C, Ma J, et al. Determination of N-methylcytisine in rat plasma by UPLC-MS/MS and its application to pharmacokinetic study. J Chromatogr B. 2015;990:118–24.
17. Pan C, Chen Y, Chen W, Zhou G, Jin L, Zheng Y, et al. Simultaneous determination of ledipasvir, sofosbuvir and its metabolite in rat plasma by UPLC-MS/MS and its application to a pharmacokinetic study. J Chromatogr B. 2016;1008:255–9
18. Wang X, Wang S, Lin F, Zhang Q, Chen HL, Wang X, et al. Pharmacokinetics and tissue distribution model of cabozantinib in rat determined by UPLC-MS/MS. J Chromatogr B. 2015;983–4:125–31.
19. Wadhwa K, Rana AC. Development and validation of HPLC-UV based bioanalytical method for the quantification of atorvastatin in rat plasma. World J Adv Res Rev. 2020;7(3):121–32.
20. Kumar S, Ali J, Baboota S. Design expert® supported optimization and predictive analysis of selegiline nanoemulsion via the olfactory region with enhanced behavioural performance in Parkinson’s disease. Nanotechnol. 2016;27(43):435101.
21. ICH harmonized guideline Q2 (R2): validation of analytical procedures. 2022 [cited 2023 Feb 05]. Available from: https://database.ich.org/sites/default/files/ICH_Q2R2_Document_Step2_Guideline_2022_0324.pdf.