
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the responsible virus causing the COVID-19 pandemic. This virus has infected a large number of people in the world, causing diseases with common cold-like symptoms to severe complications such as shortness of breath, leading to acute respiratory distress syndrome, acute respiratory failure, and death. All viruses, including SARS-CoV-2, always change along with time. Some changes in the virus structure could affect the virus’s properties, such as the ability to spreads, the disease’s severity, or the efficacy of vaccines, medicines, or diagnostic tools. SARS-CoV-2 had multiple mutations, and many variants of these viruses have emerged. The first variant (alpha) had 17 mutations and caused an increase in spreadability of 70% higher than the wild-type. Delta variant was reported to have 20 mutations. The newest variant, Omicron, shows 32 mutations, half of which reside within the spike virus’s receptor binding domain (RBD) [1–3].
SARS-CoV-2 was a positive-stranded ribonucleic acid virus with a genome that encodes structural and nonstructural proteins. Structural proteins of the virus include the spike (S), membrane (M) and envelope (E), and nucleic capsid (N) proteins. In humans, SARS-CoV-2 infection begins with the interaction of spike protein virus (S) and receptors in the host cells (angiotensin converting enzyme 2 receptor). The S protein has two functional subunits, the first is the S1 subunit mediates cell attachment, and the second is the S2 subunit which functions for the fusion of the viral and cellular membrane [4].
COVID-19 with Omicron variant cases in Indonesia was reported for the first time in December 2021 [5]. Monoclonal antibodies were one of the main treatments of COVID-19 with good efficacy clinically. Monoclonal antibodies have a unique mechanism for binding to one specific protein or receptor in the body. The targets of monoclonal antibodies against COVID-19 can be grouped into two categories. One is the antivirus antibodies, such as antibodies that target the spike protein to block viral entry. Antivirus monoclonal antibodies for treating COVID-19 include regdanvimab, bamlavimab, sotrovimab, casirivimab, and imdevimab. The second is the anti-host antibodies, such as antibodies targeting interleukin-6 receptor in the bodies to inhibit inflammation. And anti-host antibodies including tocilizumab, levilimab, sarilumab, siltuximab, canakinumab, and emapalumab [6].
Recently, regdanvimab became the only monoclonal antibody marketed in Indonesia for antivirus antibodies. Regdanvimab (CT-P59) is a recombinant human monoclonal antibody targeted against RBD of the spike protein of SARS-CoV-2. Regdanvimab (CT-P59) acts by binding the RBD of the spike protein of SARS-CoV-2 to prevent the virus from entering the cell. The efficacy of regdanvimab has been shown in treating COVID-19 patients with alpha, beta, and delta variants, both preclinical and clinically. Recent studies reported that regdanvimab (CT-P59) significantly reduced the risk of COVID-19-related hospitalization or death by 72% for patients at high risk of progressing to severe COVID-19 and 70% for all patients. Patients treated with regdanvimab (CT-P59) reported a significantly shortened time to clinical recovery by at least 4.7 days quicker for patients at high risk of progressing to severe COVID-19 and by 4.9 days quicker than placebo for all patients. Preclinical data for regdanvimab (CT-P59) also demonstrates strong neutralizing activity against the Delta variant, resulting in a 100% survival rate with virus eradication from all animals treated with regdanvimab [7].
However, the efficacy of regdanvimab for Indonesian patients with the Omicron variant has yet to be published. Clinical studies using COVID-19 patients as the subject of the study still limited to be performed in Indonesia because of the high cost of this drug. Experimental methods using X-ray crystallography, nuclear magnetic resonance (NMR), neutron diffraction, and cryo-electron microscopy has capable of generating structural models in atomic detail, but not all protein structures can be determined with these methods, and limited resources make it impossible to determine the structures of all the sequences identified in high-throughput sequencing experiments. To bridge the sequence–structure gap, computational structure prediction methods can be a solution [8].
Computational approaches such as molecular docking provide a valuable and fast alternative to experimental structural characterization for regdanvimab and RBD spike protein SARS-CoV-2 complexes. Computational approaches that can predict antibody–antigen structures would offer a valuable and fast alternative method. The residues involved in the binding interface of antibodies can be predicted quite accurately through various computational approaches [8,9].
In the present study, we investigated the binding affinity of regdanvimab to RBD SARS-CoV-2 through in silico approach, especially for the Omicron variant from an Indonesian patient. We hypothesized that regdanvimab has a high binding affinity to the RBD spike protein Omicron variant SARS-CoV-2 because regdanvimab interacts with the RBD spike protein on the conserved region of the virus. This study aimed to give initial information about the binding affinity of the regdanvimab against RBD spike protein SARS CoV-2 Omicron variant Indonesia before doing the next experimental studies in the laboratories or clinical studies.
MATERIALS AND METHODS
This study was performed in four stages: homology modeling, molecular docking, molecular dynamics (MD), and protein–protein interaction (PPI) analysis (Fig. 1) [3].
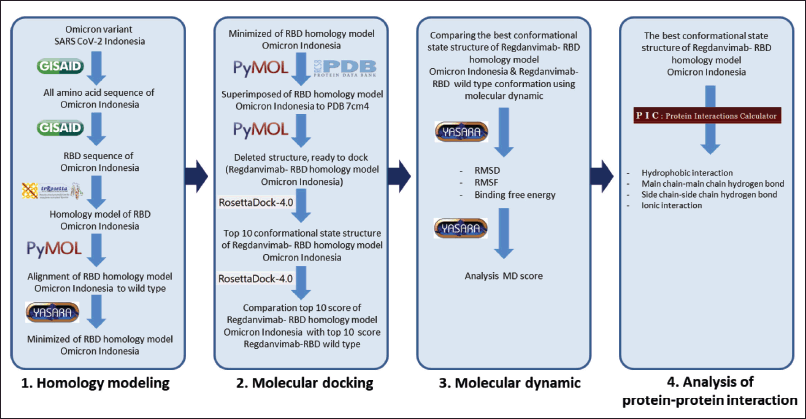 | Figure 1. The workflow of study. [Click here to view] |
This study’s homology modeling and PPI analysis were performed using a Lenovo notebook, X395, Windows 10 Pro, 64-bit operation system, Advanced Micro Devices Ryzen 5 PRO 3500U with Radeon Vega Mobile Gfx 2.10 GHz processor, 8 GB memory, and internet connection. Molecular docking and MD of this study will be used in a set of computers with Linux Ubuntu OS 20.04.5 LTS 64-bit version with Intel Core i9-10900K central processing unit (CPU) @3.70 GHz × 10 processors, 32 GB DDR4 memory, NVIDIA GeForce RTX 3090 graphic, and internet connection. The application software/platform for this study will be used software/platform of transform-restrained Rosetta (trRosetta) (web-based), RosettaDock-4.0 (web-based), Pymol, YASARA, Global Initiative on Sharing All Influenza Data (GISAID), Firefox web browser, PDF reader, and Microsoft Excel. MD will be used YASARA program; meanwhile, for analysis of PPI will be used protein interactions calculator (PIC) interface (web-based). All programs/platforms used in this study are listed in Table 1.
Homology modeling
Sequences preparation
Homology modeling aims to predict a structure from its sequence with an accuracy comparable to experimentally achieved best results. In this case, no NMR or crystalized structure of RBD Omicron Indonesia exists in the Protein Data Bank database. We use homology modeling to make a three-dimensions (3-D) RBD Omicron Indonesia structure based on the amino acid sequence obtained from the GISAID database. We selected the sample of Omicron Indonesia from the GISAID database with accession number EPI_ISL_13501802. This sample was the newest patient on the GISAID database when we retrieved it on July 2nd, 2022. To identify the mutation, we performed alignment of the amino acid sequences of the Omicron variant from an Indonesian patient with a wild-type virus from Wuhan as a reference using CoVsurver from GISAID [10]. Thereafter, we separated RBD sequences only as the initial data to perform homology modeling using the trRosetta web server platform.
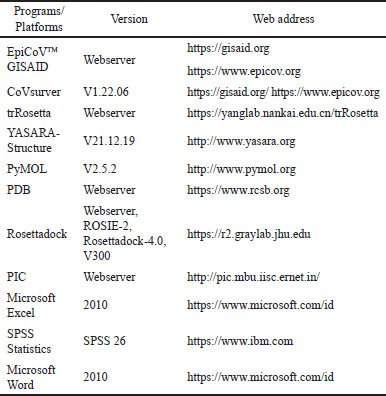 | Table 1. Programs used in this in silico study. [Click here to view] |
Running trRosetta webserver
The trRosetta server is a web-based platform powered by deep learning and Rosetta for fast and accurate protein structure prediction. The RBD structure is located in the virus spike between Thr333 and Gly526 residues [3]. We edited the amino acid sequences using Microsoft Word to get these RBD sequences. To perform homology modeling using the trRosetta webserver, we must upload and submit the RBD Omicron variant sequence from Indonesia. The result of the homology modeling from trRosetta would be generated within less than 1 hour.
The advantage of trRosetta programs from other similar structure prediction servers is a web-based platform with rapid and accurate de novo structure prediction supported by deep learning and Rosetta. In general, it only takes about 1 hour to predict the final structure of a typical protein with a length of 300 amino acids because trRosetta is supported by a cluster system with 10 CPU cores working in parallel. This procedure is carried out by entering the amino acid sequence of a protein. Then a deep neural algorithm is used first to predict the geometry of the residues, including their distances and orientations. The geometric prediction results are then transformed as restraints to guide the prediction structure based on direct energy minimization applied in the Rosetta framework [11].
Alignment using the Pymol program
After getting the result of the 3-D structure from homology modeling, we aligned and visualized the RBD model 3-D structure Omicron Indonesia to the RBD crystal structure of the wild-type to confirm that our homology model has a similar crystal structure. We used protein data bank (PDB) number 7CM4 as a reference to do this alignment. The alignment and visualization were performed using the Pymol program. We used the RMSD score to analyze the results of the alignment. The lower the RMSD score, the better the accuracy of the result [12].
Minimized energy using the YASARA program
The next step was to perform energy minimization of the homology model structure to confirm that there was no clash, bad contact, and geometry in the structure and to obtain the lowest energy of the structure [13]. In energy minimization of homology models, the smaller the RMSD to the experimental structure, the better the energy landscape suited for getting there. We performed energy minimization using the YASARA program. To perform energy minimization in the YASARA program, we need to set the force field environment using AMBER14. In this case, we set on default temperature & pH. The energy minimization was conducted using the macro-Em_runclean.mcr script. And then, the minimized structure needs re-align with the crystal structure and visualized using the Pymol program [14].
Molecular docking
Structure preparation
After confirming that the RBD structure of the homology model has a high similarity with the crystal structure, the next stage was performed molecular docking of RBD Omicron Indonesia to the regdanvimab structure. We used the regdanvimab structure from PDB number 7CM4. To perform molecular docking, we used a Rosettadock-4.0 webserver (Rosie) (https://r2.graylab.jhu.edu). RosettaDock was the top-performing method for computational protein-protein docking [15–17]. Molecular docking for this study will be used a homology model of RBD spike protein SARS-CoV-2 Omicron variant structure from Indonesia that the sequence was obtained from GISAID (https://gisaid.org) and the regdanvimab structure obtained from Protein Data Bank (www.rcsb.org).
To perform molecular docking, we must prepare the initial binding poses of the complex structure of regdanvimab-RBD Omicron Indonesia. The initial binding poses were generated by superimposing the RBD Omicron Indonesia structure on a regdanvimab-RBD wild-type crystal structure obtained from PDB 7CM4. And then RBD wild-type chain (chain A) from PDB 7CM4 were deleted. Any unwanted water and residue molecules were deleted. We got the initial binding poses of the complex structure of regdanvimab-RBD Omicron Indonesia which is ready to perform molecular docking using the Rosettadock-4.0 webserver (Rosie) [18–20].
Running molecular docking
After the structure complex is prepared, we upload and submit the structure to the Rosettadock-4.0 webserver. We generated the docking simulations using motif dock score as a centroid score function with the Rosetta application number V300. We also performed a docking simulation of the PDB number 7CM4 structure as a reference. In the Rosetta-4.0, local docking performs perturbation of ~ ±3 Å in the direction between the two proteins, ~8 Å in the directions sliding the proteins relative to each other along their surfaces, ~8° of tilt of the proteins, and a complete 360° spin around the axis between the centers of the two proteins. The server will generate 1,000 independent simulations from this range of random positions. Swaps are also performed with conformations from the pregenerated ensemble to accommodate for backbone flexibility in docking [19].
In Rosetta’s docking, the binding score was used interface score to rank the complexes generated from molecular docking. Rosetta interface score is defined as Isc = Ebound−Eunbound, where Ebound was the score of the bound complex, and Eunbound was the sum of the scores of the individual protein partners in isolation. To predict the possible neutralizing antibody of regdanvimab to SARS-CoV-2, the interface binding scores were compared between regdanvimab-RBD Omicron Indonesia (homology modeling) and regdanvimab-RBD wild-type (crystal structure). The statistical significance was tested using an independent t-test [21].
Rosettadock-4.0 will generate 1,000 binding poses of the complex structure during molecular docking, resulting in different interface binding scores. The best binding pose of the complex is the structure with the lowest interface binding score. In this study, to analyze the binding affinity, we compared the interface binding score (I_sc) between the regdanvimab-RBD Omicron Indonesia structure and reganvimab-RBD wild-type crystal structure from PDB 7CM4 as reference [3,21].
Molecular dynamics
MD simulations were performed to confirm possible interactions between regdanvimab and RBD spike protein SARS CoV-2 Omicron variant Indonesia and to visualize its conformational changes in micro or nanosecond perturbation. The advantages of MD simulations are that it gives a route to the system’s dynamical properties, transport coefficients, time-dependent responses to perturbations, and rheological properties [22].
In this study, the binding affinities of the regdanvimab-RBD Omicron Indonesia were also predicted using MD. The best structure was generated from molecular docking, then performed MD to confirm that this structure and binding affinity would not change significantly on dynamic simulations [3]. MD simulations of this study were performed using the YASARA V21.12.19 program.
Ligand-protein complexes were prepared before simulation, using the best conformational structure from molecular docking. The simulation cell boundary is set to periodic. Atoms that stick out of the simulation cell will be wrapped to the opposite side of the cell during the simulation. The simulation was carried out in an explicit water environment, at constant pressure, using an AMBER14 force field, in a periodic cell boundary condition, and the model was simulated at 298 K (25ºC). Force field energies help to analyze the structural quality of a protein, and distortions of local covalent geometry can be analyzed by looking at the bond, angle, and planarity energies. The MD simulation was performed for 20 ns with 201 snapshots generated in every 100 ps using the md_run.mcr script in YASARA 21.12.19.L.64. The RMSD and root mean square fluctuation (RMSF) values were analyzed for each simulation using the md_analyze.mcr script, and also free energy of binding were analyzed using the Rosetta score on the YASARA structure of MD. The visualization was displayed using YASARA and PyMOL.
The equilibrium states were considered reached if the average deviation of the backbone atoms’ root-mean-squared deviations (RMSD) value was less than 1 Å [23–26]. To analyze the binding affinity, we compared RMSD and RMSF variation between the regdanvimab-RBD Omicron Indonesia structure and reganvimab-RBD wild-type crystal structure generated from MD. We used Microsoft Excel to perform the mean calculation of RMSD.
To determine the interaction between regdanvimab and RBD Omicron Indonesia, the interface binding score energy between regdanvimab and RBD Omicron Indonesia was calculated using the Rosetta score and compared to regdanvimab-RBD wild-type structure resulting from MD. The binding free energy of each snapshot of the regdanvimab-RBD binding in the last 5 ns of the MD simulations was calculated using the Rosetta score on the YASARA structure.
Analysis of PPI
PPI analysis was essential in understanding the molecular basis of stability and functions of the proteins. Several interactions determine the stability of a protein structure. PIC interface was a server that, given the coordinate set of the 3-D structure of a protein or an assembly, computes various interactions such as disulfide bonds, interactions between hydrophobic residues, ionic interactions, hydrogen bonds, aromatic–aromatic interactions, aromatic–sulfur interactions, and cation–n interactions within a protein or between proteins in a complex. PIC interface also aids in recognizing and analyzing various interactions in tertiary structures of proteins and in recognizing interacting motifs that are exposed or buried. The advantage of the PIC server is the easy availability of inter-residue interaction calculations in a single site [27].
We also performed PPI analysis to describe PPI in the regdanvimab-RBD Omicron Indonesia structure. This study used the PIC interface platform to do this PPI analysis (http://pic.mbu.iisc.ernet.in/) [27]. We compared the PPI between the regdanvimab-RBD Omicron Indonesia structure and regdanvimab-RBD wild-type crystal structure, especially for the hydrophobic interaction, main chain-main chain hydrogen bonds, main chain-side chain hydrogen bonds, side chain-side chain hydrogen bonds, ionic interactions, aromatic-aromatic interactions, aromatic-sulfur interactions, and Pi-cation interactions [28]. We used Microsoft Excel to analyze and compare the data of interface residues of regdanvimab-RBD Omicron Indonesia from the PIC interface.
RESULT AND DISCUSSIONS
Homology modeling
To identify the mutations, we performed amino acid alignment of the Omicron variant Indonesia with a wild-type virus from Wuhan using the CoVsurver platform from GISAID. We found that the similarity of both sequences was 97.3% [10]. For the mutations, the data showed that the Omicron variant from Indonesian patients has 34 mutations in the spike protein, including 17 mutations in the RBD region (Fig. 2). The number of these mutations was quite different from the Omicron variant from South Africa published by Hwang et al. [3] which has 32 mutations, including 15 mutations in the RBD regions. These mutations would affect the characteristics of the Omicron Indonesia variant virus [10].
 | Figure 2. Mutations of the amino acid sequence of Omicron Indonesia compared to the wild-type. Underline letters: mutations that increased the binding affinity of the virus to hACE2 [3]. Blue letters: mutations in the RBD region of SARS-CoV-2 Omicron Indonesia compared to RBD wild-type [10]. [Click here to view] |
 | Figure 3. RBD sequences of Omicron variant Indonesia. Blue letters: mutations in the RBD of SARS CoV-2 Omicron Indonesia [10]. Red letters: The RBD structure was between the Thr333 and Gly526 residues [3]. [Click here to view] |
The pictures of the RBD amino acid sequences of Omicron variant Indonesia obtained from GISAID with accession number EPI_ISL_13501802 that has been edited using Microsoft Word can be seen in Figure 3.
After the RDB sequences were obtained from GISAID, we used this sequence to perform homology modeling using the trRosetta webserver. The results of trRosetta showed that they generated five model structures with the highest score, and the best model of these five models has a template modeling (TM)-score of 0.753 (Fig. 4). The TM-score is defined to assess the topological similarity of two protein structures:
where L is the length of the target protein, and Lali is the number of equivalent residues in two proteins. di is the distance of the ith pair of the equivalent residues between the two structures, which depends on the superposition matrix; max means the procedure to identify the optimal superposition matrix that maximizes the sum in the equation. The scale d0 is defined to normalize the TM-score in a method where the magnitude of the average TM-score for random protein pairs is independent of the size of the proteins. TM-score stays in (0, 1) with a higher value indicating a stronger similarity. TM-score can be used as an approximate but quantitative criterion for protein topology classification; i.e., protein pairs with a TM-score >0.5 are mostly in the same fold, while those with a TM-score <0.5 are mainly not in the same fold. In this study, the homology model of RBD Omicron Indonesia has a TM-score of 0.752 that indicates a good result [29].
In the next step, we performed alignment of the RBD model structure Omicron Indonesia to the RBD crystal structure of wild-type from PDB number 7CM4 using the Pymol program. The result showed that the RMSD score from alignment was 1.176 Å; this means that our homology model is not significantly different from the crystal structure (Fig. 5). Yeni and Tjahyono [12], stated that the goal of RMSD for homology modeling is less than 2 Å. This showed that our homology model of RBD Omicron Indonesia was a good model.
To confirm that there was no clash in the structure and got the lowest energy of the structure, we performed energy minimization of the homology model using the YASARA Structure program. In energy minimization, the smaller the RMSD from the initial structure, the closer the model is to reality [14]. The minimized structure was then re-aligned with the crystal structure and visualized using the Pymol program. The RMSD result of the minimized structure still shows a low score (RMSD 1.664 Å). It shows that the resulting 3-D structure of homology modeling RBD Omicron Indonesia does not significantly differ from the 3-D structure of RBD crystal structure after energy minimization [12]. This result of the minimization model becomes the final model for performing molecular docking.
Molecular docking
In this study, we performed molecular docking of RBD Omicron Indonesia to the regdanvimab structure from PDB 7CM4 using the Rosettadock-4.0 webserver (Rosie). RosettaDock has been among the top-performing methods for computational protein–protein docking. RosettaDock was a multiscale Monte Carlo-based docking algorithm that utilized a centroid-based coarse grain stage to quickly identify favorable docking poses and an all atom refinement stage that simultaneously optimized rigid-body position and side-chain conformation. The initial binding poses were prepared for structure by superimposing the RBD Omicron Indonesia structure on a regdanvimab-RBD wild-type crystal structure obtained from PDB 7CM4. Thereafter RBD wild-type chain from PDB 7CM4 was deleted. As a result, we got the initial binding poses of the complex structure of regdanvimab-RBD Omicron Indonesia that is ready to perform molecular docking using a Rosettadock-4.0 webserver (Fig. 6a).
Rosettadock-4.0 generated 1,000 binding poses of the complex structure in molecular docking. The best binding pose is the structure with the lowest interface binding score. In this case, we used the top 10 conformations to analyze and compare the docking score between regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type (Fig. 6b). The resulting plot of interface binding score versus RMSD can be seen in Figures 7 and 8. In this study, to analyze binding affinity, we compared the interface binding score (I_sc) of the regdanvimab-RBD Omicron Indonesia structure with the reganvimab-RBD wild-type crystal structure from PDB 7CM4 as a reference [3,21]. The table shows a comparison of the top 10 interface binding scores of both structures in molecular docking (Table 2).
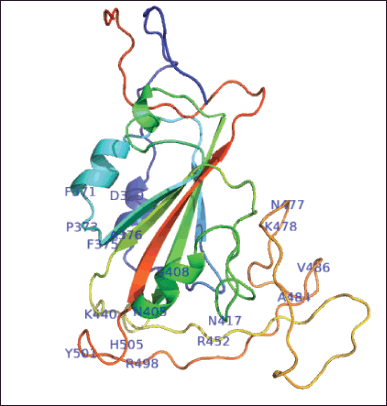 | Figure 4. Structure 3-D homology modeling of RBD SARS CoV-2 Omicron Indonesia using trRosetta. The label shows the mutations of the RBD sequences. [Click here to view] |
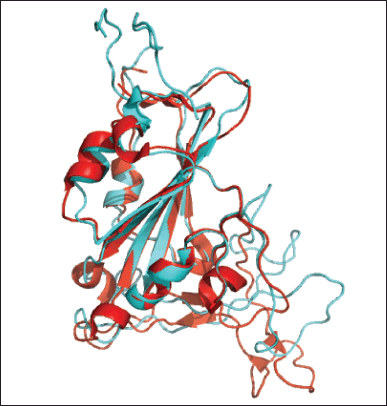 | Figure 5. Alignment of RBD Omicron Indonesia structure (cyan) with RBD wild type crystal structure (red) using Pymol program. RMSD: 1,176 Å. [Click here to view] |
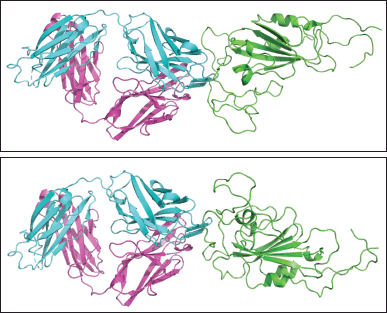 | Figure 6. (a). Initial binding pose of regdanvimab-RBD Omicron Indonesia structure before docking. Green: RBD Omicron Indonesia; cyan: heavy chain of regdanvimab; purple: light chain of regdanvimab. (b). The best binding pose of regdanvimab-RBD Omicron Indonesia structure with the lowest interface binding score. Green: RBD Omicron Indonesia; cyan: heavy chain regdanvimab; purple: light chain regdanvimab. [Click here to view] |
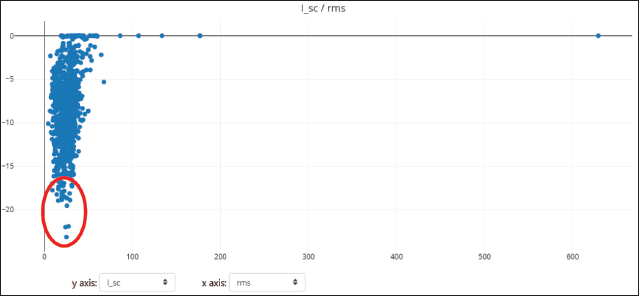 | Figure 7. Plots of 1,000 docking conformations generated by Rosettadock-4.0 for regdanvimab-RBD Omicron Indonesia. Dots composed by interface binding score (I_sc) versus RMSD. Red circle: top 10 models. [Click here to view] |
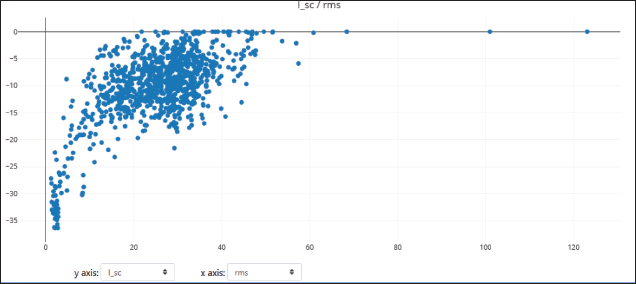 | Figure 8. Plots of 1,000 docking conformations generated by Rosettadock-4.0 for regdanvimab-RBD wild type. Dots composed by interface binding score (I_sc) versus RMSD. [Click here to view] |
The data showed that the interface binding score of regdanvimab-RBD Omicron Indonesia structure was significantly higher than that of regdanvimab-RBD wild-type (−19.881 ± 1.74 vs. −35.097 ± 1.01 kcal/mol respectively, p < 0.05, independent t-test). In the Rosetta binding score, the lower the score, the better the binding. These results indicated that the binding affinity of regdanvimab-RBD Omicron Indonesia was lower than that of regdanvimab-RBD wild-type. These studies were in line with the study published by Hwang et al. [3] which stated that the binding affinity of the Omicron variant to regdanvimab was significantly weaker than that of the wild-type. However, the data showed that the binding affinity of regdanvimab-RBD Omicron Indonesia still exists but with a lower affinity. According to Ye et al. [30], structures with interface scores lower than −9.0 were classified as complexes with good binding affinity.
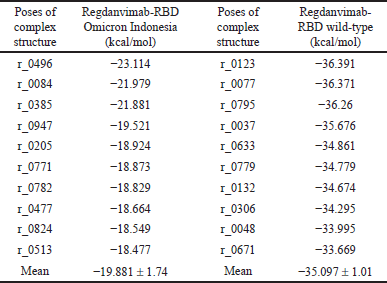 | Table 2. Top 10 interface binding score (I_sc) from molecular docking using Rosettadock-4.0 webserver. [Click here to view] |
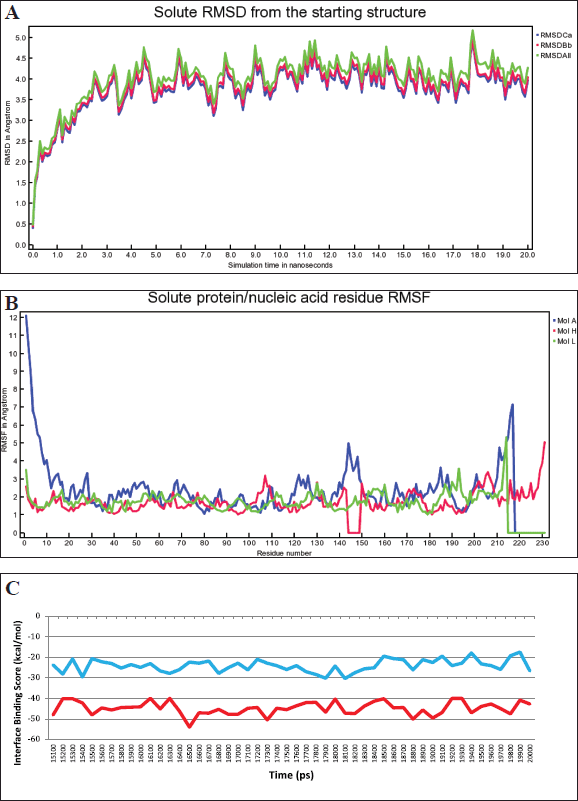 | Figure 9. (a). RMSD changes in the complex of regdanvimab-RBD Omicron Indonesia during MD simulations (RMSDCa, C alpha RMSD; RMSDBb, backbone RMSD; RMSDAll, all-heavy atom RMSD). (b). RMSF value of the complex of regdanvimab-RBD Omicron Indonesia during MD simulations (Mol A, RBD SARS-CoV-2 Omicron Indonesia; Mol H, heavy chain of regdanvimab; Mol L, light chain of regdanvimab). (c). Interface binding score energy of regdanvimab-RBD Omicron Indonesia (blue) and regdanvimab-RBD wild-type (red) during the last 5 ns of MD simulations. [Click here to view] |
Molecular dynamics
The binding affinities of the regdanvimab-RBD Omicron Indonesia were also predicted using MD. The best structure generated from molecular docking was used as the initial conformations and then MD was performed using the YASARA program. MD simulations were performed over 20 ns. The results of MD of the regdanvimab-RBD Omicron Indonesia showed that RMSD, RMSF, and binding free energy can be seen in Figure 9a–c. This study’s RMSD value was relatively flat and stable during the MD simulation of regdanvimab-RBD Omicron Indonesia. The stabilization seems significant after the first 3 ns. The structure of the regdanvimab-RBD Omicron was considered stable if the deviation of the RMSD values of at least for the last 5 ns duration of the MD simulations was less than or equal to 1 Å [23–26]. The stability of the regdanvimab-RBD Omicron Indonesia structure during MD simulations for 20 ns was shown as a good result. The delta of RMSD on the last 5 ns was 0.18 Å (Table 3). Analysis of backbone RMSD on the last 5 ns showed that regdanvimab can stabilize RBD Omicron Indonesia because regdanvimab can decrease the deviations of the atoms significantly. This can be seen from the RMSD variability shows less than 1 Å in the last 5 ns in both regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type.
During these MD simulations, we also monitored the RMSF value. The greater the RMSF value of amino acids, the more flexible they are in the binding process [13,30]. The RMSF of the regdanvimab-RBD Omicron Indonesia structures in this study was also plotted as a function of residue (Fig. 9b). The regions where regdanvimab has a large impact on RBD Omicron Indonesia residue sequences were also evaluated by RMSF analysis. The RMSF values of the 211–217 (529–535) residues of the A chain (RBD Omicron Indonesia) and the 214 residues of the L chain (regdanvimab) fluctuated significantly in the process of binding between regdanvimab and the RBD Omicron Indonesia as presented in Figure 9b. Especially for the RMSF values of residue number 217 of the A chain (Lys529) reached 7.13 Å showing the flexible analysis at the RBD Omicron Indonesia tail [31].
For further determination of the interaction between regdanvimab and RBD Omicron Indonesia, the binding free energy (interface binding score) between regdanvimab and RBD Omicron Indonesia was calculated using the Rosetta score and compared to regdanvimab-RBD wild-type. MD simulation combined with binding free energy calculation has been applied widely to study the interaction between drugs and targets. These steps calculate the binding free energy difference between bound and unbound state structures in the receptor-ligand systems. The binding free energy was calculated when the simulation reached equilibrium states [26]. In this case, we used interface score energy from Rosetta scoring on the YASARA structure to analyze binding free energy. The results showed that regdanvimab-RBD Omicron Indonesia produced a higher level of interface binding score energy in the last 5 ns than regdanvimab-RBD wild-type, that is −25.13 ± 3.06 versus −44.69 ± 3.15 kcal/mol, respectively (p < 0.05, independent t-test) (Fig. 9c). These values indicate that regdanvimab-RBD Omicron Indonesia has lower binding affinity than regdanvimab-RBD wild-type.
 | Table 3. Backbone RMSD on the last 5 ns during MD simulations of regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type. [Click here to view] |
Analysis of PPI
We also performed PPI analysis using the PIC interface platform to describe PPI in the regdanvimab-RBD Omicron Indonesia structure [27]. We found hydrophobic interaction on Y449, Y451, Y495, and Y501 RBD Omicron Indonesia structure residues with regdanvimab. The number of these hydrophobic interactions was lower than that on the RBD wild-type structure. We also found that the main chain-main chain hydrogen bond occurs on the S494 residue of RBD Omicron Indonesia, the same interaction as that on the RBD wild-type structure. For ionic interaction, we found different interactions between RBD Omicron Indonesia and wild-type complexes on R452, R466 residues, and R403, E484 residues, respectively. The differences in interfacial contact residues between regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type can be seen in Table 4. The data from the alignment of the amino acid sequences of the SARS-CoV-2 Omicron variant from Indonesia shows that 17 point mutations occur in the RBD region. When we analyzed these mutations, 3 of 17 point mutations occurred on the binding interface region between regdanvimab and RBD Omicron Indonesia (L452R, Q498R, and N501Y). These mutations were suggested to cause the decreased binding affinity of the regdanvimab-RBD Omicron Indonesia. We can conclude that from these changes in the PPI, the binding affinity of the regdanvimab-RBD Omicron Indonesia was lower than that regdanvimab-RBD wild-type
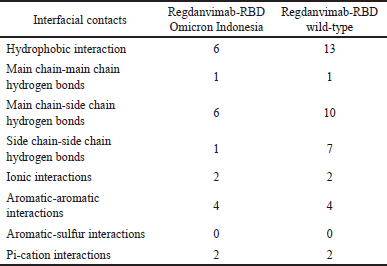 | Table 4. Differences in interfacial contacts between regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type. [Click here to view] |
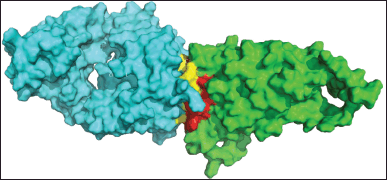 | Figure 10. Interface interaction of regdanvimab (cyan) and RBD Omicron Indonesia (green). [Click here to view] |
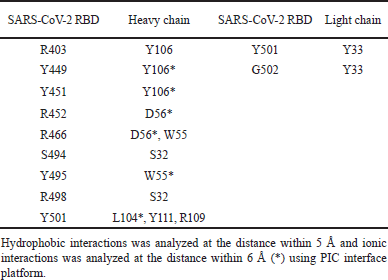 | Table 5. Interacting residues between regdanvimab and SARS-CoV-2 RBD Omicron Indonesia. [Click here to view] |
 | Table 6. Interacting residues between regdanvimab and SARS-CoV-2 RBD wild-type [32] . [Click here to view] |
The interface interaction between regdanvimab and RBD Omicron Indonesia is depicted in Figure 10. The complete PPI between regdanvimab-RBD Omicron Indonesia and regdanvimab-RBD wild-type can be seen in Tables 5 and 6, respectively.
CONCLUSION
This study found that the Omicron variant of SARS-CoV-2 from Indonesian patients has 34 mutations, including 17 mutations within the RBD region. From the amino acid sequence of this Omicron variant, we made homology modeling of RBD Omicron Indonesia using the trRosetta webserver with TM-score 0.753. The homology model also showed a good result after the energy-minimizing step using the YASARA program and alignment using the Pymol program. In the molecular docking step of the regdanvimab-RBD Omicron Indonesia complex structure using Rosettadock-4.0 webserver in this study, we found that the binding affinity of regdanvimab-RBD Omicron Indonesia was lower than that of regdanvimab-RBD wild-type as reference. After performing MD of the regdanvimab-RBD Omicron Indonesia complex structure using the YASARA program, we found that the result of RMDS showed that the complex structure has good stability, as indicated by the delta RMSD for the last 5 ns was less than 1 Å. And after investigating the PPI using the PIC interface, we found that the number of residues involved in the binding interface of the regdanvimab-RBD Omicron Indonesia structure was lower than that of the regdanvimab-RBD wild-type structure. Based on the results of this in silico study, molecular docking, MD, and PPI analysis, it can be concluded that the binding affinity of regdanvimab-RBD Omicron Indonesia structure was lower than that of regdanvimab-RBD wild-type structure. To validate this finding, further in vitro and in vivo studies are needed to know the efficacy of regdanvimab in treating COVID-19 with the Omicron variant in Indonesia.
LIST OF ABBREVIATIONS
3-D, Three-dimensions; AMD: Advanced Micro Devices; CPU, Central processing unit; GISAID, Global Initiative on Sharing All Influenza Data; NMR, Nuclear magnetic resonance; PDB, Protein data bank; PIC, Protein interaction calculator; RBD, Receptor binding domain; RMSD, Root mean square deviation; RMSF, Root mean square fluctuation; SARS-CoV-2, Severe Acute Respiratory Syndrome Corona Virus 2 ; TM-score: template modeling score.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the facilities and materials provided by Dexa Laboratories of Biomolecular Sciences. The authors would also thank Destrina Grace Simanjuntak and Gerardine Emeralda Andyahesti for proofreading the manuscript.
AUTHORS’ CONTRIBUTIONS
L.B.I. designed the methodology, collected data, analyzed data, and prepared the manuscript. R.R.T put forward the idea and supervised this research. V.D.P. contributed to the review of the manuscript.
FINANCIAL SUPPORT
This research was funded by internal funding from PT. Dexa Medica.
CONFLICT OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Verma J, Subbarao N. Insilico study on the effect of SARS-CoV-2 RBD hotspot mutants’ interaction with ACE2 to understand the binding affinity and stability. Virology. 2021;561:107–16. doi: https://doi.org/10.1016/j.virol.2021.06.009
2. Hurt AC, Wheatley AK. Neutralizing antibody therapeutics for COVID-19. Viruses. 2021;628(13):1–15. doi: https://doi.org/10.3390/v13040628
3. Hwang S, Baek SH, Park D. Interaction analysis of the spike protein of delta and Omicron variants of SARS-CoV?2 with hACE2 and eight monoclonal antibodies using the fragment molecular orbital method. J Chem Inf Model. 2022;62:1771–82. doi: https://doi.org/10.1021/acs.jcim.2c00100
4. Jahanshahlu L, Rezaei N. Monoclonal antibody as a potential anti-COVID-19. Biomed Pharmacother. 2020;129:1–4. doi: https://doi.org/10.1016/j.biopha.2020.110337
5. Kementrian Kesehatan Republik Indonesia [Internet]. Kasus Pertama Omicron di Indonesia Diduga dari WNI yang Datang dari Nigeria. Available from: https://sehatnegeriku.kemkes.go.id/baca/rilis-media/20211219/5339013/kasus-pertama-omicron-di-indonesia-diduga-dari-wni-yang-datang-dari-nigeria/ (Accessed 12 February 2023)
6. Ning L, Abagna HB, Jiang Q, Liu S, Huang J. Development and application of therapeutic antibodies against COVID-19. Int J Biol Sci. 2021;17(6):1486–96. doi: https://doi.org/10.7150/ijbs.59149
7. Celltrion Healthcare Co.,Ltd. [Internet]. Summary of product characteristic. Regkirona. Available from: https://www.ema.europa.eu/en/documents/product-information/regkirona-epar-product-information_en.pdf’ (Accessed 12 February 2023).
8. Weitzner BD, Jeliazkov JR, Lyskof S. Modeling and docking of antibody structures with Rosetta. Nat Protoc. 2017;12(2):401–16. doi: https://doi.org/10.1038/nprot.2016.180
9. Ambrosetti F, Garcia BJ, Touris JR, Bonvin AMJJ. Modeling antibody-antigen complexes by information-driven docking. Structure. 2019;28:119–29. doi: https://doi.org/10.1016/j.str.2019.10.011
10. Global Initiative on Sharing Avian Influenza Data [Internet]. EpiCoVTM. Available from: https://www.epicov.org/epi3/frontend#3731b2 (Accessed 20 June 2022).
11. Du Z, Su H, Wang W. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc. 2021;16:5634–51. doi: https://doi.org/10.1038/s41596-021-00628-9
12. Yeni Y, Tjahyono DH. Homology modeling of isocitrate dehydrogenase type 1 (R132H) epitopes using modeller, I-Tasser and (PS)2 for glioma vaccine. Farmasains. 2017;4(1):21–32.
13. Abusham RAK, Masomian M, Salleh AB, Leow ATC, Rahman RNZR. An in-silico approach to understanding the structure-function: a molecular dynamics simulation study of rand serine protease properties from Bacillus subtilis in aqueous solvents. Adv Biotechnol Microbiol. 2019;12(2):9–17. doi: https://doi.org/10.19080/AIBM.2019.12.555828
14. Krieger E. YASARA science manual. Nijmegen, The Netherlands: Frick Digitaldruck; 2017. 1–100 pp.
15. Gray JJ, Moughon S, Wang C, Kuhlman OSB, Rohl CA, Baker D. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–99. doi: https://doi.org/10.1016/S0022-2836(03)00670-3
16. Daily MD, Masica D, Sivasubramanian A, Somarouthu S, Gray JJ. CAPRI rounds 3–5 reveal promising successes and future challenges for RosettaDock. Proteins. 2005;60:181–86. doi: https://doi.org/10.1002/prot.20555
17. Chaudhury S, Sircar A, Sivasubramanian A, Berrondo M, Gray JJ. Incorporating biochemical information and backbone flexibility in RosettaDock for CAPRI rounds 6–12. Proteins. 2007;69:793–800. doi: https://doi.org/10.1002/prot.21731
18. Lyskov S, Gray JJ. The RosettaDock server for local protein–protein docking. Nucleic Acid Res. 2008;36:W233–8. doi: https://doi.org/10.1093/nar/gkn216
19. Chaudhury S, Berrondo M, Weitzner BD. Benchmarking and analysis of protein docking performance in Rosetta v3.2. Plos One. 2011;6(8):e22477. doi: https://doi.org/10.1371/journal.pone.0022477
20. Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, et al. Serverification of molecular modeling applications: the Rosetta online server that includes everyone (ROSIE). Plos One. 2013;8(5):e63906. doi: https://doi.org/10.1371/journal.pone.0063906
21. Park T, Lee SY, Kim S, Kim MJ, Kim HG, Jun S, et al. Spike protein binding prediction with neutralizing antibodies of SARS-CoV-2. BioRxiv. 2020;1–22. doi: https://doi.org/10.1101/2020.02.22.951178. Available from: https://www.biorxiv.org/content/10.1101/2020.02.22.951178v1.article-info
22. Allen MP. Introduction to molecular dynamic. In: Attig N, Binder K, Grubmuller H, Kremer K, editors. Computational soft matter: from synthetic polymers to proteins. Julich, Germany: NIC Series; 2004. vol. 23, pp 1–28.
23. Wicaksono RG, Hariono M, Istyastono EP. Molecular dynamics studies of full human matrix metalloproteinase 9 liganded with N-hydroxy-2-[(4-phenylphenyl) Sulfonyl-propan-2-yloxyamino] acetamide. J Farm Sains Komun. 2020;17(1):1–7. doi: https://doi.org/10.24071/jpsc.001970
24. Prasasty VD, Istyastono EP. Short communication: structure-based design and molecular dynamics simulations of pentapeptide AEYTR as a potential acetylcholinesterase inhibitor. Indones J Chem. 2020;20(4):953–59. doi: https://doi.org/10.22146/ijc.46329
25. Istiyastono EP. Molecular dynamics studies of matrix metalloprotenase-9 stabilization by caffeic acid. J Farm Galenika (Galenika J Pharm). 2020;6(2):280–85. doi: https://doi.org/10.22487/j24428744.2020.v6.i2.15013
26. Liu X, Shi D, Zhou S, Liu H, Liu H, Yao X. Molecular dynamics simulations and novel drug discovery. Expert Opin Drug Discov. 2017;13(1):1–15. doi: https://doi.org/10.1080/17460441.2018.1403419
27. Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35:W473–6. doi: https://doi.org/10.1093/nar/gkm423
28. Shishir TA, Jannat T, Naser IB. An in-silico study of the mutation-associated effects on the spike protein of SARS-CoV-2, Omicron variant. PLoS One. 2022;17(4):e0266844. doi: https://doi.org/10.1371/journal.pone.0266844
29. Xu J, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Struct Bioinform. 2010;26(7):889–95. doi: https://doi.org/10.1093/bioinformatics/btq066
30. Ye C, Hu W, Gaeta B. Prediction of antibody-antigen binding via machine learning: development of data sets and evaluation of methods. JMIR Bioinform Biotech. 2022;3(1):e29404. doi: https://doi.org/10.2196/29404
31. Zhang L, Wang P, Yang Z, Du F, Li Z, Wu C. Molecular dynamics simulation exploration of the interaction between curcumin and myosin combined with the results of spectroscopy techniques. Food Hydrocoll. 2020;101:1–9. doi: https://doi.org/10.1016/j.foodhyd.2019.105455
32. Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021;12:288. doi: https://doi.org/10.1038/s41467-020-20602-5