
INTRODUCTION
Drug discovery and development are lengthy and extremely costly. Many existing molecules can be delivered to be new drugs. Nevertheless, it is practically impossible to do in wet-lab experiments. On the other hand, available computational capacity has given way to new methodologies in silico to screen extensive drug libraries. The interest in applying machine learning (ML) techniques as drug design tools has grown over the last decades (Gertrudes et al., 2012). Advancements in computational science have accelerated drug discovery and development. This step prior to preclinical investigations lowers the economic cost and expands the scope for future drug discovery. ML techniques have gained significant prominence in the pharmaceutical industry, offering the ability to accelerate and automate the analysis of a large amount of available data (Carracedo-Reboredo et al., 2021; Patel et al., 2020).
Virtual screening (VS) has emerged as an essential tool in drug development because it conducts efficient in silico searches across millions of compounds, resulting in higher yields of potential drug leads. VS is a computational method to find structural candidate compounds, drug design, and molecular modeling by screening a chemical compound database (Carpenter & Huang, 2018; Vyas et al., 2008). It is widely used in the early stage drug discovery and development process. Hermansyah et al. (2021) created a VS methodology that used a ML-based quantitative structure-activity relationship (QSAR) strategy to screen millions of compounds for the DPP-4 inhibitor. Another popular in silico method for VS was molecular docking, which could be used to investigate the interactions between related proteins and small compounds or peptides in cancer inhibition (Widyananda et al., 2021). It combines the various methods available to find a list of hit and lead compounds that meet the criteria for proceeding to the proof through the wet laboratory.
Liver cancer is the 6th most common cancer, with 905,677 (4.7%) new cases in 2020 for both sexes and all ages. It is the fourth leading cause of death (830,190 deaths, or 8.3% of all deaths), as published by the International Agency for Research on Cancer in 2020 (GLOBOCAN 2020). Hepatocellular carcinoma (HCC) accounts for 85 to 90% of cancer deaths in primary liver cancers. With high-quality screening, and careful management of screen-discovered lesions, appropriate for the disease stage, HCC can be prevented, detected early, and effectively treated (Marrero et al., 2018). Drug research for liver cancer therapy was discovered and developed from active compounds of medicinal plants (Kaushik et al., 2020; Manosroi et al., 2015). Herbals include many biologically active compounds showing promising antiproliferative activities against HCC. Phytochemicals were consumed as dietary agents or supplements since they provide beneficial health effects in adjuvant therapy or chemoprevention (Ranzato et al., 2014; Vidya Priyadarsini & Nagini, 2012). Several drugs, such as chemotherapy agents, were used to treat liver cancer, which often causes side effects and cancer resistance.
The in vitro assay uses hepatoma cell lines, habitually applied and tailored as a model, to assess the molecular subtype-specific medication response giving precision therapy for HCC patients (Hirschfield et al., 2018). Due to the heterogeneity of gene mutations and the complex and diverse molecular pathogenesis of HCC, molecular targeting therapy for HCC is difficult (Bai et al., 2020). When performing an efficacy study, hepatoma cell lines, HepG2 cells, are frequently used as in vitro models to examine drugs’ uptake, metabolism, excretion, and toxicity due to the limited availability of fresh human hepatocytes (Wi?niewski et al., 2016). The advantage of using hepatocellular carcinoma cells is that they are highly available as an immortalized cell line, which can be preserved and easily handled. The study highlighted that metabolizing enzyme activity from HepG2 cells is not reduced in culture, as occurs in primary cultures of human hepatocytes. HepG2 cells express many differentiated hepatic functions, such as lipoprotein and triglyceride metabolism, glycogen and bile acid synthesis, and secretion of plasma proteins and cholesterol or insulin signaling. They also keep the morphological characteristics of normal liver cells (Donato et al., 2015; González et al., 2017).
This study aims to apply an ML model as a VS tool for bioactive compounds in plants that predict potential antiproliferative activity against HCC. This activity can reduce the number of compounds tested in the wet lab using the HepG2 cell model as an in vitro model. Thus this activity results in the efficiency of time, cost, and effort in discovering anticancer drugs.
EXPERIMENTAL
Material and methods
This research used AMD Ryzen 9 5900HX 3.3 GHz with 32 GB RAM. The ML model has been built with three critical stages, including data acquisition, selection of fingerprint features, building ML model, and VS stages. This stage, especially in the first and second stages, was built referring to the Automated QSAR modeling made by Kausar (2018) and Hermansyah (2021). In this experiment, we added a VS stage to obtain chemical compounds from herbal bioactive compounds using fingerprint features and the best-performing ML model that has been built. This model was created using a workflow from the Konstanz Information Miner (KNIME) version 4.3.4 software (https://www.knime.com), the most comprehensive free platform for open-source ML applications that can be used for modeling and visualization with no coding required to be able to perform (Berthold et al., 2008; Hermansyah et al., 2021). The conceptual framework for ML for antiproliferative VS of herbal compounds is seen in Figure 1.
Dataset of antiproliferative activity against human HepG2 cells
ChEMBL database with target Homo sapiens HepG2 cells and IC50 activity was used as a dataset (https://www.ebi.ac.uk/chembl/target_report_card/CHEMBL395/). The dataset of 21,345 molecules has been filtered by eliminating salt molecules and duplicate data, leaving 5,460 molecules with antiproliferative activity in nanomolar (nM) as the activity unit. Normalized data of activities (IC50) in the nanomolar unit is converted into a negative value algorithm in molar units (pIC50 = 9-log (IC50). The data are labeled based on the pIC50 value, in which pIC50 > 6.0 is labeled as active, pIC50 < 5.0 is labeled as inactive, and the rest is unlabeled data. There are 851 active records, 3,089 inactive records, and 1,520 unlabeled records. This value will be used as a reference target for classification and prediction in the ML algorithm. Three thousand one hundred fifty-two data were used as data learning, and 788 data were used as data testing (external test). A thousand five hundred twenty unlabeled data points were removed.
Data curation and featured selection
Filter out unneeded data columns that only have molecule name, IC50 standard, and SMILES present. Each fingerprint was calculated or described based on RDKit, CDK, and fingerprint bit expansion. ML will use this value as a reference target for classification and regression. Random partitioning was performed at 80:20 for data learning and testing.
ML model selection and optimization
The ML models will also be chosen, including random forest, deep learning, XG Boost, SVC, and decision tree. Each ML model is seen for its performance from the internal and external validation, made with classification and regression models.
The performance of the regression model can be seen from the value of precision and accuracy, while in the regression model, it can be seen from the value of R2 and MSE. The internal test uses the ten-fold cross-validation method with 80% of the total data, or 3152 data, while the external test uses 20% or 788 data. Its optimization using hyperparameter tuning was done to maximize the performance value of each ML model. The best ML models were performed for further VS.
Virtual screening
An ML model demonstrating the best performance was used to perform a VS of bioactive compounds in plants. The screening was carried out on 3,608 bioactive compounds from several medicinal plants that had been collected based on SMILES and selected fingerprint representation. These molecules were tested using the ML-based QSAR classification model to determine their activity. The active molecule was further processed for similarity assay. The activity of molecules that meet the similarity criteria was determined further using the QSAR regression model to predict compounds’ IC50 against HepG2 cell antiproliferation. In VS, several stages were carried out to find the most potent compound through a similarity test with Tanimoto coefficient value above 0.8, the activity having a pIC50 value above six and meeting at least three Lipinski drug-like rules.
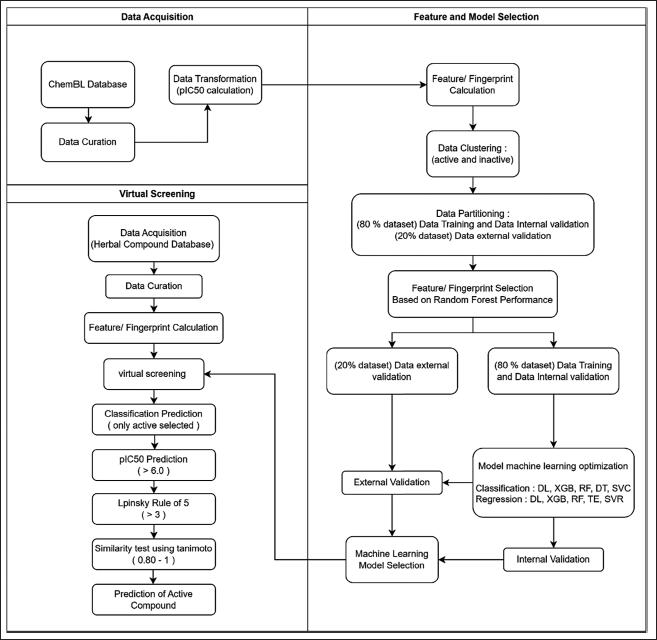 | Figure 1. Conceptual framework for ML for VS. [Click here to view] |
RESULTS AND DISCUSSION
The development of new high-tech systems for screening anticancer drugs is one of the main problems of preclinical screening. Significant money, time, and several stages are required to find and develop new medications as anticancer agents. Many failures occurred at each stage due to the compounds tested failing to pass the stage, such as in efficacy, safety, and ADME properties. The poor correlation between preclinical in vitro and in vivo data and clinical trials needs improvement, and the expansion of new high-tech preclinical in vitro screening systems is becoming very important (Kitaeva et al., 2020).
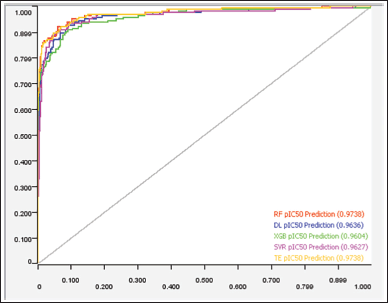 | Figure 2. RoC graph supports the performance results of ML regression models (Hermansyah, 2021). [Click here to view] |
ChEMBL is a free, large-scale bioactivity database that was mainly collected from the medicinal chemistry literature manually. It consists of information on the compounds examined (including their structures), the biological or physicochemical assays performed on them, and the targets of these assays in a structured way (Bento et al., 2014). In this study, the data learning is collected and filtered from the ChEMBL database that results from in vitro testing which used Homo sapiens HepG2 cell lines and their IC50 value as antiproliferative activity. ML based on QSAR assumed that structurally similar compounds have corresponding bioactivity properties. The ML model used this approach to virtual screen bioactive compounds for predicting, identifying, or discovering novel drug candidates. MLing-based VS is a computational technique used for screening large datasets of molecules and has been successfully used to complement HTS for drug discovery.
Molecular fingerprints are used to describe mathematical objects by representing molecule structures. The fingerprints employed in this work are divided into rule-based, applying binary value categories. Many fingerprints address an alternate part of the particle, which can enormously influence search execution (Cereto-Massagué et al., 2015). Different kinds of fingerprints encode various aspects of the molecules (Muegge & Mukherjee, 2016). The most common rule-based fingerprints are MACCS structural keys, which depend on molecular topology. In a predefined atom neighborhood, circular topological fingerprints represent a mixture of nonhydrogen atom types and routes. In addition, pharmacophore fingerprints contain regional properties linked to molecular identification (Zagidullin et al., 2021). Because of its remarkable effectiveness in molecular structure comparisons requiring the identification of compounds with similar bioactivity, ECFP is frequently utilized. Circular fingerprints are known to have several valuable properties, including the ability to be calculated quickly, the capacity to represent virtually infinitely many different molecular features, including stereochemical information, the presence of specific substructures represented by their features, and the ability to adapt the ECFP algorithm to be applied to other works (Rogers & Hahn, 2010).
Fingerprints are numerical variations of n components (bits) in length, where n is typically between 166 and 1024. Many kinds of fingerprints address an alternate part of the particle, which can enormously influence search execution (Cereto-Massagué et al., 2015) and encode various aspects of molecules (Muegge & Mukherjee, 2016).
The random forest was utilized as the initial benchmark to pick and determine the feature or fingerprint representation of molecules because of the tree-based tactics used, which naturally rank by how effectively they improve the purity of the node. This implies a reduction in impurity across all trees. Nodes with the most significant reduction in impurity occur at the beginning of the trees, while notes with the smallest reduction in impurity occur towards the end of the trees. Thus, we can create a subset of essential features by pruning trees below a particular node.
The selected fingerprints were then tested using a regression and classification model. The best performance results from the two models are then used as a feature for optimizing and selecting ML models (Table 1).
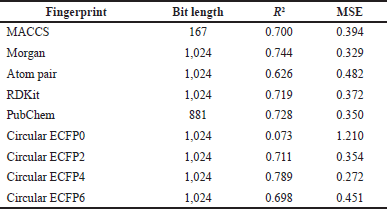 | Table 1. Regression performances of various fingerprints. [Click here to view] |
Experimental results show that the Circular Fingerprint ECFP4 has the best consistency in both models (Table 2). The performance of R2 and MSE of the regression model is 0.789 and 0.272, respectively. While the fingerprint circular ECFP4 classification model also gets the best performance with accuracy and precision, respectively, at 0.954 and 0.942. Different fingerprints showed an ability to explain organic molecules with small sizes and extended connectivity fingerprints with up to four bonds (ECFP4), representing the molecule better than other fingerprints studied. ECFP4 is used as molecular structure descriptors for comparing ML algorithms in antiproliferative activity against HepG2 cell lines as in vitro HCC model.
A total of five ML models have been tested as ML models for antiproliferative HCC. Three thousand one hundred fifty-two pieces of learning data, 788 pieces of testing data, and 1,024 pieces of feature data were used to select and optimize the model. The performance of this ML model has been tested internally and externally and created using ML types of classification and regression models. Some ML models have been performed to predict the activity and were constructed using ECFP4, representing molecule structures, and pIC50 as proliferative activity values.
The experiments in the ML classification model showed that random forest performed better on internal and external validation, with accuracy values of 0.936 and 0.954, respectively. This is also supported by the precision results on internal and external tests, 0.912 and 0.950, respectively.
Similar to the classification model, random forest is still
better in the regression model compared to the four other models (Figure 2 and Table 5). The random forest regression values for internal and external validation are 0.746 and 0.803, respectively. The ML random forest model was the best model with the results obtained. It was chosen as a virtual model for screening chemical compounds in plants for antiproliferative HCC.
The best hyperparameters tuning for the classification ML model is 189 number model and 171 tree depth for the random forest, 491 number of rounds and 184 threads for XGBoost, 7 Node, and 357 Threads for the decision tree, 587 costs and 75 degrees for SVC, two dense layers, ADAM, stochastic gradient descent, RELU weight, 326 batch size and 435 epoch for deep learning. The best hyperparameters tuning for regression ML model is 732 tree depth and 500 number of models for the random forest, 74 batch size and 312 epoch for deep learning, 31 boosting rounds, 0.171 etas, nine max depth for XGBoost, 376 costs and 437 degrees for support vector regression and 672 number models for tree ensemble.
Random forest was used as a supervised ML algorithm for the VS of bioactive compounds in plants. It creates decision trees from various samples, using the majority vote for classification and the average for regression. This technique is appropriate for the VS of a vast compound library to identify molecules as active or inactive or to rank them according to their activity levels. After going through VS, several medicinal plant compounds are predicted to have high activity by looking at their pIC50 values. With the Tanimoto value of 1.0, it can be ascertained that the compound is identical to the control compound (active compound classified from ChemBL395 data). Meanwhile, compounds with a Tanimoto value below 1.0 mean that these compounds are not identical and can be categorized as new types of compounds that can be submitted as references as active ingredients for the search for new drugs.
The Tanimoto coefficient is considered very good and recommended to be used as a screening method for the similarity of a compound. However, this is used only to ensure that the compound is similar to the active compound from the ChemBL395 dataset for antiproliferative HepG2. Even without using Tanimoto, the 257 samples that are predicted to be active can be accounted for because the level of accuracy of ML produced in this study is above 90% (Tables 3 and 4).
It was found that 66 samples had a Tanimoto index above 0.80 and 49 compounds had pIC50 predicted values above 6.0. Only 15 compounds passed by fulfilling 3 if investigated using Lipinski’s rules. Figure 3 shows six molecules and their predicted activities.
Virtually screening compounds using this ML model obtained compounds that are potentially antiproliferative HepG2 cells in vitro. These results align with previous studies that indicated that this plant has activity as an anticancer of the liver. Zerumbone prominently performed an antiproliferative activity toward HepG2 cells with an IC50 of 3.45 ± 0.026 ug/mL (Lv et al., 2018) and 23.64 ± 1.23 µM (Sakinah et al., 2007). At the same time, antiproliferative activity against HepG2 cells of Tylophorine was observed with IC50 11 ± 4 nm (Gao et al., 2004). Gossypol, a natural compound derived from cottonseed, was identified as an inhibitor of some cancer cell lines (Lan et al., 2015; Xu et al., 2019; Yu et al., 2020). Antofine, isotylocrobrine, and 6’-hydroxy-3,4,2’,3’,4’-pentamethoxychalcone have not been previously investigated in detail in any other pharmaceutical applications. Thus, this current study proposes that those compounds might be used as a starting point to be applied for new antiproliferative drugs.
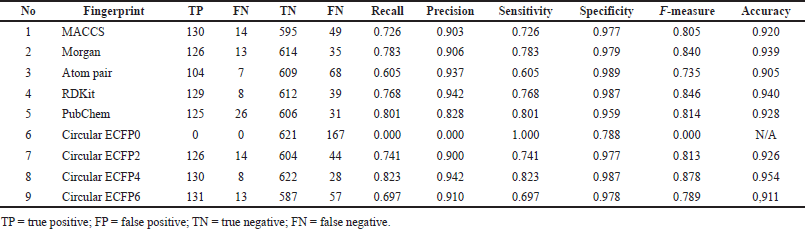 | Table 2. Classification performances of various fingerprints. [Click here to view] |
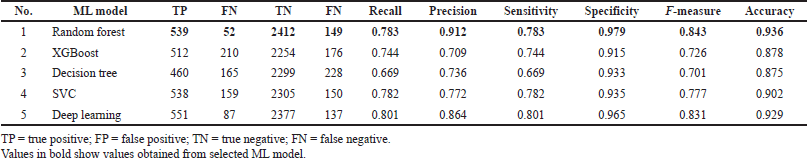 | Table 3. Classification performance of internal validation. [Click here to view] |
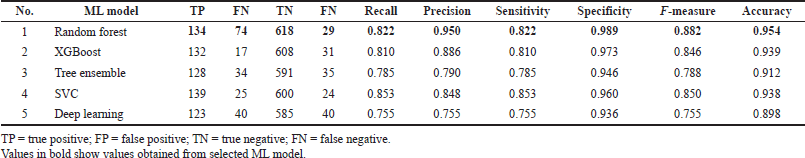 | Table 4. Classification performance of external validation. [Click here to view] |
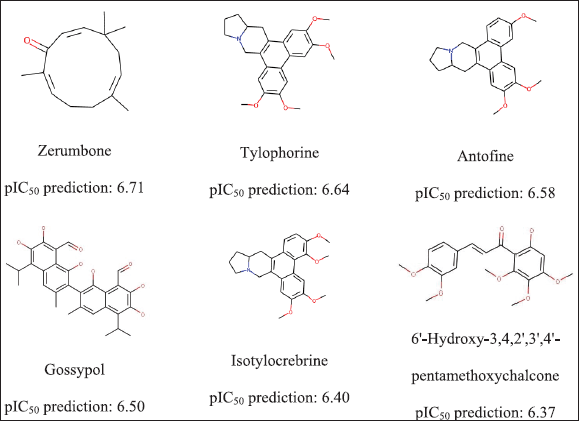 | Figure 3. Active prediction of HepG2 antiproliferative compounds. [Click here to view] |
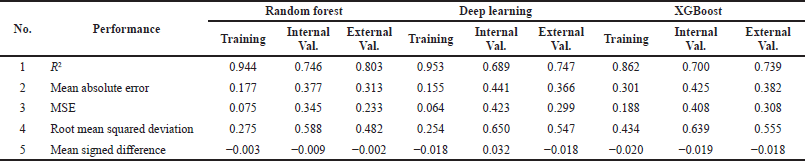 | Table 5. Regression performance (Hermansyah, 2021). [Click here to view] |
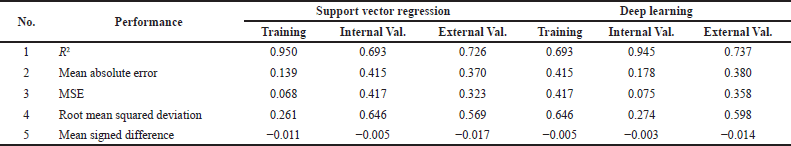 | Table 5. Regression performance (Hermansyah, 2021). (Continued) [Click here to view] |
CONCLUSION
The ML model could be performed as a VS of bioactive compounds that predict potential antiproliferative activities against HCC. In our studies, random forest algorithm has suitable performance constructed using ECFP4 molecule structures representation. Application of this model makes it much easier to screen the molecular structure that has potential efficacy for a specific target with safety and minimum environmental or health impacts. In this approach, the software is utilized to build a higher number of possible chemical structures, which are then used to forecast qualities relating to performance, safety, health, and environmental impact based on QSAR. The bioactive compounds that predict activities could be developed as drug candidates and tested in wet laboratories.
ACKNOWLEDGMENTS
This research received funding from Organization Research for Life Science and Environment, BRIN no.9/III/HK/2022.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Bai L, Ren Y, Cui T. Overexpression of CDCA5, KIF4A, TPX2, and FOXM1 coregulated cell cycle and promoted hepatocellular carcinoma development. J Comput Biol, 2020; 27(6):965–74 CrossRef
Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP. The ChEMBL bioactivity database: an update. Nucleic Acid Res, 2014; 42(D1):D1083–90. CrossRef
Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B. Studies in classification, data analysis, and knowledge organization, 2008; (pp. 319–26). Springer Berlin, Heidelberg, Germany. CrossRef
Carpenter KA, Huang X. Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Design, 2018; 24(28):3347–58. CrossRef
Carracedo-Reboredo P, Liñares-Blanco J, Rodríguez-Fernández N, Cedrón F, Novoa FJ, Carballal A, Maojo V, Pazos A, Fernandez-Lozano C. A review on machine learning approaches and trends in drug discovery. Comput Struc Biotechnol J, 2021; 19:4538–58. CrossRef
Cereto-Massagué A, Ojeda MJ, VallsC, Mulero M, Garcia-Vallvé S, Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods, 2015; 71(C):58–63. CrossRef
Donato MT, Tolosa L, Gómez-Lechón MJ. Culture and functional characterization of human hepatoma HepG2 Cells. Protocols in vitro hepatocyte research. Springer, New York, NY, pp. 77–93, 2015. CrossRef
Gao V, Lam W, Zhong S, Kaczmarek C, Baker DC, Cheng YC. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res, 2004; 64(2):678–88doi:10.1158/0008-5472.CAN-03-1904. CrossRef
Gertrudes JC, MaltarolloVG, Silva RA, Oliveira PR, Honorio KM, Da Silva ABF. Curr Med Chem, 2012; 19(25):4289–97. CrossRef
GLOBOCAN 2020. Mortality. Globocan 2020.
González LT, Minsky NW, Espinosa LEM, Aranda RS, Meseguer JP, Pérez PC. In vitro assessment of hepatoprotective agents against damage induced by acetaminophen and CCl4. BMC Complement Alternat Med, 2017; 17(1):39. CrossRef
Hermansyah O, Bustamam AA. and Yanuar A. Virtual screening of dipeptidyl peptidase-4 inhibitors using quantitative structure-activity relationship-based artificial intelligence and molecular docking of hit compounds. Comput Biol Chem, 2021; 95:107597 CrossRef
Hirschfield H, Bian CB, Higashi T, Nakagawa S, Zeleke TZ, Nair VD, Fuchs BC, Hoshida Y. Experiment Mol Med, 2018; 50(1):e419. CrossRef
Kausar S, Falcao AO. An automated framework for QSAR model building. J Cheminformat, 2018; 10(1):1. CrossRef
Kaushik N, Yang H, Jeong SR, Kaushik NK, Bhartiya P, Nguyen LN, Choi EH, Kim JH. Biological and medical applications of plasma-activated media, water and solutions. Appl Sci (Switzerland), 2020; 10(21):1–6. CrossRef
Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV. Cell culture based in vitro test systems for anticancer drug screening. Front Bioeng Biotechnol, 2020; 8:322. CrossRef
Lan L, Appelman C, Smith AR, Yu J, Larsen S, Marquez RT, Liu H, Wu X, Gao P, Roy A, Anbanandam A, Gowthaman R, Karanicolas J, De Guzman RN, Rogers S, Aubé J, Ji M, Cohen RS, Neufeld KL, Xu L. Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol Oncol, 2015; 9(7):1406–20. CrossRef
Lv T, Zhang W, Han X. Zerumbone suppresses the potential of growth and metastasis in hepatoma HepG2 cells via the MAPK signaling pathway. Oncol Lett, 2018; 15(5):7603–10. CrossRef
Manosroi A, Akazawa H, Kitdamrongtham W, Akihisa T, Manosroi W, Manosroi J. Potent antiproliferative effect on liver cancer of medicinal plants selected from the Thai/Lanna medicinal plant recipe database “MANOSROI III”. Evid Based Complement Alternat Med. 2015;2015:397181. CrossRef
Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, Roberts LR, Heimbach JK. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology, 2018; 68(2):723–50. CrossRef
Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov, 2016; 11(2):137–48. CrossRef
Patel L, Shukla T, Huang X, Ussery DW, Wang S. Machine learning methods in drug discovery. Molecules, 2020; 25(22):5277. CrossRef
Ranzato E, Martinotti S, Calabrese CM, and Calabrese GG. Role of nutraceuticals in cancer therapy. J Food Res, 2014; 3(4):18. CrossRef
Rogers D, Hahn MM. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J Chem Inform Model, 2010; 50(5):742–54. CrossRef
Zhang S, Liu Q, Liu Y, Qiao H, Liu Y. Zerumbone, a Southeast Asian Ginger Sesquiterpene, Induced Apoptosis of Pancreatic Carcinoma Cells through p53 Signaling Pathway. Evid Based Complement Alternat Med. 2012;2012:936030. CrossRef
Priyadarsini VR, Nagini S. Cancer chemoprevention by dietary phytochemicals: promises and pitfalls. Curr Pharm Biotechnol, 2012; 13(1):125–36. CrossRef
Vyas V, Jain A, Gupta A. Virtual screening: a fast tool for drug design. Sci Pharm, 2008; 76(3):333–60. CrossRef
Widyananda MH, Pratama SK, Samoedra RS, Sari FN, Kharisma VD, Ansori ANM, Antonius Y. Molecular docking of anthocyanins and ternatin in Clitoria ternatea as coronavirus disease oral manifestation therapy. J Pharm Pharm Res, 2021; 9(4):484–96. CrossRef
Wi?niewski JR, Vildhede A, Norén A, Artursson P. In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J Proteomics, 2016; 36:234–47. CrossRef
Xu J, Zhu GY, Cao D, Pan H, Li YW. Gossypol overcomes EGFR-TKIs resistance in non-small cell lung cancer cells by targeting YAP/TAZ and EGFRL858R/T790M. Biomed Pharmacother, 2019; 115:108860. CrossRef
Yu Q, Hu Z, Shen Y, Jiang Y, Pan P, Hou T, Pan ZQ, Huang J, Sun Y. Gossypol inhibits cullin neddylation by targeting SAG-CUL5 and RBX1-CUL1 complexes. Neoplasia (United States), 2020; 22(4), 179–91. CrossRef
Zagidullin B, Wang Z, Guan Y, Pitkänen E. Comparative analysis of molecular fingerprints in prediction of drug combination effects. Briefings Bioinform, 2021; 22(6):bbab291. CrossRef