
INTRODUCTION
Bioanalytical techniques are applied to measure and target biological compounds or drugs and its components in biological fluids such as blood, serum or plasma, urine, and gastric content. These techniques are precise and highly useful and recorded for therapeutic drug monitoring (TDM) (Ermer and Miller, 2006; Viswanathan et al., 2007). In clinical practice, the quantitative bioanalysis of drugs and their metabolites provides an important new approach to personalized medicine. It helps the clinician to individualize drug treatment for drugs characterized by a short therapeutic range, and/or decrease the risk of dose-dependent adverse effects (Cremers et al., 2016). In addition, the newly available TDM approach has led to several advances that aimed at measuring drug concentrations and relating these results to therapeutic efficacy or secondary effects. Nowadays, several analytical methods were used in routine clinical laboratories, including automated immunoassays (AI), high performance liquid chromatography (HPLC), and liquid chromatography tandem-mass spectrometry (LC-MS/MS) (Mou and Jiang, 2017; Vogeser and Kirchhoff, 2011). However, for such screening in routine bioassay, thin-layer chromatography, gas chromatography (GC) with currently used detectors, and HPLC with a diode array detector (DAD) are mostly applied, but GC-MS is by far the most widely used method in this setting. Although GC-MS is widely applied as the primary reference method for bioanalysis, the intensive work and time-consuming procedures limit their use in the clinical laboratory. On the other hand, the spectrum is largely used in clinical practice, a simple but less selective method. AI is simple to perform with minimal sample preparation and faster (turnaround time). However, it is well recognized that AI is sensitive to cross-reactivity and cannot measure different substances simultaneously. Following analytical approaches, there are significant variations between AI methods in terms of patient outcomes as well as reference areas; these differences are responsible for the sensitivity of the antibodies to interference or cross-reaction with structurally similar compounds (Horie et al., 2007; Lee et al., 2006; Taieb et al., 2002). A such bioanalytical validation procedure should support the strength and significance of outcoming results (Boulanger et al., 2003; Tiwari and Tiwari, 2010). The initial validation is the first step which must be continuously monitored during the application process to prove its performance (Riley and Rosanske, 1996). Some studies have discussed biomedical validation steps and described protocol for an effective strategy (Ermer and Miller, 2006; Tiwari and Tiwari, 2010). However, bioanalytical method validations require an appropriate statistical analysis to evaluate precision, analytical range, accuracy, sensitivity/specificity, and limit of quantification (LOQ) and detection (LOD). These steps must follow a practical protocol and the obtained results should be compared with predefined quality criteria (Food and Drug Administration, 2016; Peters and Maurer, 2002). According to high importance of bioanalysis assay validation in the field of TDM, some guidance documents related have been published (Capiau et al., 2019; van Nuland et al., 2020). There is a permanent need for reliable and thoroughly validated bioanalytical techniques in order to detect and measure drugs and other substances in complex mixtures of compounds, such as drugs compositions as well as their metabolites in biological matrix. For several years, the TDM represents an important approach to optimizing clinical care that facilitates the clinician to individualize the treatment in relationship with the patient’s physiological profile, control drug doses to achieve a systemic level associated with the desired therapeutic goal, and also reduce the risk of dose-dependent adverse effects (Cremers et al., 2016). The role of many TDM assay laboratories is to quantify the concentrations of drugs in a sample and relate this concentration to a therapeutic level published in the literature. The aim of this review was to resume different criteria of bioanalytical method validation and their use in the clinical studies in TDM relation. It was mainly focused on the validation of chromatographic methods that are mostly used. This review paper has also discussed application of solving routine problems related to validation process.
REGULATION PROCEDURES
Previously, bioanalysts operated under a single Food and Drug Administration (FDA) bioanalytical method validation guideline, but the background differs in regulatory vocabulary that could influence the routine practice of numerous bioanalytical laboratories. The first bioanalytical guideline was published by the FDA (2001). Following this guideline, several draft guidelines have been published in the area of bioequivalence/bioavailability guidelines is indicated in Table 1, which offer only a brief overview of recommended necessary bioanalytical steps or a recommendation to further bioanalytical standards. The operator is then responsible for using the alternative guidelines to provide complete operational requirements and processes for the performance of the analytical techniques. In this regard, a comprehensive supplementary guidance project has been issued in 2009 from the European Medicines Agency (EMA) (Whitmire et al., 2011), which also outlines the requirements for bioanalytical guidance and approaches that parallel those outlined in the workshop papers (Shah et al., 2000; Viswanathan et al., 2007) or FDA guidance (FDA, 2001). In bioanalytical method validation process, EMA and FDA guidelines are widely used despite some differences in the requirements of each guideline. The guideline provided by the EMA and the FDA on bioanalytical technique validation is largely comparable but not similar. On the basis of the above, there are some differences in the recommended validation parameters. We generally found the format of the FDA guideline clearer and the tables in its supplement very useful. The EMA provides a more precise description of the practical performance of trials. The FDA presents the reports more comprehensively. For bioanalytical method such as liquid chromatography, we also found that the International Conference on Harmonisation (ICH) to be very practical because it combines the advantages of both EMA and FDA guidance to reduce terminology confusion and unnecessary effort to comply with two or more guidelines.
METHOD DEVELOPMENT
The development of bioanalytical method needs the evaluation and optimization of the different steps such as sample preparation, chromatographic separation, detection systems, quantification, matrix effects and stability of chemical compounds and drugs in the biological matrix.
Sample preparation
Biological matrix in general, such as blood, serum/plasma, and urine, due to their complexity and protein concentration, is not suited for direct injection in bioanalytical equipment. A preanalytical step therefore is crucial to prepare the material for the bioanalytical technique (Ashri and Abdel-Rehim, 2011; Nováková and Vl?ková, 2009). The objective of sample preparation is to eliminate interfering compounds (including proteins, salts, and lipids) and also to concentrate the analytes. Due to the various physicochemical features of these drugs, selecting the best sample preparation provides a difficulty to methods that quantify drug concentrations.
The most typical extraction techniques presented in Figure 1 are liquid-liquid extraction (LLE) (Blanchard et al., 1988; Chang et al., 2007; Remane et al., 2010), solid-phase extraction (SPE) (Dunér et al., 2007; Poole, 2003), and protein precipitation (PP) (Burgess, 2009; Chang et al., 2007; Souverain et al., 2004). However, during the development of bioanalytical method, the PP and LLE are the major sample preparation techniques for bioanalysis using LC-MS (Ali et al., 2008; Raynie, 2006), whereas the PP was made by adding of a precipitating solvent to biological samples using organic solvents such as methanol (MeOH), acetonitrile (ACN), or trichloroacetic acid. In this practical method, the chemical agent used in the sample preparation decreases the plasma’s dielectric constant, which improves the attractivity of proteins, resulting in precipitation and protein accumulation (Ryan, 2011). However, LLE showed in Figure 2 may come with some limitations. In case of multiple analytes extraction, it is important to verify that they all have similar partition ratios (similar polarity), as the recovery will not be equivalent. In general, SPE, LLE, and PP are widely applied for sample preparation and enrichment of analytes in biological matrix in many bioanalytical laboratories has been summarized in Table 2. Based on previously published research and our own scientific expertise, sample preparation procedures should be identified and improved depending on the objective of bioanalytical method. A suitable technique must be chosen in relation to extraction time, selectivity, number of steps, solvent consumption, and the ability to organize on-line techniques. In this context, sample preparation is frequently the most difficult aspect of developing a bioanalytical procedure. In addition to the above-mentioned sample preparation procedure, affinity chromatography is the single technique that allows purification of an analyte on the basis of biological function or specific chemical structure; this chromatographic technique plays a unique and significant role in separation technology. The use of this separation method allows for highly selective extraction of the target and structurally comparable substances (e.g., a drug and its metabolites) from heterogeneous matrices. However, the materials used to create this specific preparation sample were frequently cheap, quick, and reproducible; they also had a high capacity and could be recycled and used multiple times.
 | Table 1. Some international bioanalytical guidance available for bioanalytical method validation. [Click here to view] |
 | Figure 1. Case of PP protocol for plasma samples using organic solvents. [Click here to view] |
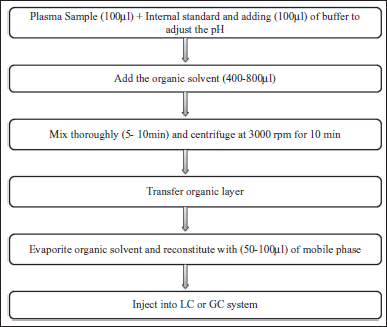 | Figure 2. Example for LLE steps for plasma samples. [Click here to view] |
Detectors
For selectivity and sensitivity in the step of bioanalytical method development, the selection of detection tools is extremely important. In some cases, for bioanalysis drugs and their metabolites or biological compounds, methods using ultraviolet DAD (UV-DAD), UV, and mass spectrometry (MS) detector have been applied in the literature. The UV-DAD is an appropriate detector for the identification of this class of drugs and allows a high level of sensitivity for polyunsaturated species. However, UV detection does not distinguish between compounds with similar chromophore groups. More detailed structural information can be taken when an MS is coupled to a UV-DAD. MS is a useful detection for qualitative analysis to identify and confirm the molecular patterns of unknown drugs and is particularly suitable of its high selectivity and sensitivity (Lazaou et al., 2000). The benefits of combining liquid chromatography with LC-MS or LC-MS-MS have been demonstrated for many analyses used in various bioanalyses of drugs and their metabolites (Ackermann et al., 2002; Lee and Kerns, 1999). The components of an LC-MS system comprise the autosampler, the LC apparatus, and the ionization source that is a part of the mass spectrometer that ionizes compounds and mass spectrometer. In the majority, these components are managed by a single software package. It can be noted that for interfacing LC with MS, there are certain limitations on the mobile phases and flow rate that can be used. The most common solvents of the mobile phase are chemical agents applied to the mobile phase that are used for the chromatographic separation of the sample analysis. During the method development, the impact of small changes in the ratio of solvent making up the mobile phase and buffer pH will influence the peak resolution of analytes.
Typical reversed-phase LC systems connected to the MS may use a combination of water/methanol or ACN for the mobile phase. There are restrictions on the components of mobile phase for example, it should be volatile. Typical mobile phase may also contain ammonium acetate, acetic acid, and formic acid. Many papers are available which focus on LC parameters that seem useful in LC-MS analysis (Ackermann et al., 2002; Hsieh et al., 2003; Tiller et al., 2003). In fact, LC-MS and LC-MS-MS are used for measurements of newly synthesized substances that are part of a library of compounds. These techniques evaluate that the appropriate product was synthesized and that the purity is adequate for usage in the library. In a subsequent stage, LC-MS is used to examine different physical and chemical characteristics (such as physiological solubility, permeability, and chemical stability) of these new compounds. In addition, a variety of drug metabolite and pharmacokinetic (PK) assays are used in drug discovery to measure the properties of the PK stage of drug molecule, as well as their PK parameters. Several of these tests are dependent on LC-MS or LC-MS-MS for the measurement stage (Tiller et al., 2003).
 | Table 2. Comparison of three sample preparation methods (PP, LLE, and SPE). [Click here to view] |
Selectivity
During bioanalytical method development steps, the selectivity parameter is a critical criterion for the drug assay and other compounds in biological matrix. This bioanalytical norm is defined as the capacity to detect analyte concentration without interference from sample components; the selectivity for bioanalytical methods must be determined with respect to metabolites of endogenous compounds and known degradation products prior to the validation process. Presumably, the interference merely exists in a trace form and can have a negative effect on the quantification of unidentified compound at concentration approaching to the quantification limit (Vessman, 1996). Selectivity for interference from endogenous compounds in biological matrices could be determined by treating a minimum of diverse sources providing the same blank matrices (Valcárcel et al., 2001). A careful analysis of chromatograms over interest peak time windows is necessary to assess selectivity; it must be highlighted that it is not suitable to try a single source of blank matrix (Hartmann et al., 1998). However, it is better to test the used blanks and they need to be free from any noise or interference. On the other hand, factors such as the subject’s consumption of food and drinks, the ingestion of vitamins additive, the use of nonprescription and prescription drugs other than those tested, and smoking may affect the selectivity criteria. When selecting an inaccurate detection system, serious problems may arise especially when analyte metabolites are undetectable or there are no identifiable known degradation products. In this case, it is necessary to perform the synthesis of specific degradation products and known metabolites, if possible, to validate to verify selection. In the absence of control sample for metabolites or breakdown products, the assays described below may be sufficient to validate selectivity. Biological samples from the patients under treatment could be the best solution; these specimens should be assayed according to the usual chromatographic requirements under varying chromatographic parameters in order to address a large number of potential concerns combining peaks sampling (Dadgar and Burnett, 1995; Peters and Maurer, 2002). If concentrations are sufficiently high and the UV spectrum of the potential byproducts or intermediates change from those of the parent molecule, to provide a purity peak, other multiwavelength detectors, such as a diode array, can be used.
Analyte stability
All aspects of analyte stability should be clearly defined: patient preparation, sample collection, transportation to laboratory, handling of the sample in laboratory including storage, and stability during all stages of pretreatment (i.e., stability in an organic solvent). Moreover, according to symposium report (Shah et al., 2000), the stability of the analyte was determined as follows: the chemical stability of an analyte in a particular matrix, under specific conditions and for specific time intervals. Generally, there are two types of stability: (1) the stability of the analyte during the different stages of pretreatment and (2) the analyte stability in biological sample. The analyte stability depends on its physicochemical characteristics and the conditions of storage and conservation. The stock solutions’ stability and individual analytes must be tested under normal laboratory conditions of temperature, humidity, and light for at least 6 hours in comparison with extemporaneously prepared solution. The storage conditions of these solutions must also be clearly established (4°C, −20°C, etc.). Furthermore, the stability of the analytes must be verified during all stages of pretreatment of the sample (stability in the organic solvent, in the dry extracts, on the automatic sample changer, etc.). In pure solutions, the analyte and the internal standard (IS) are considered stable if the deviation from the theoretical concentration does not exceed 2%. A difference up to 5% is tolerated for the stability tests in the dry extracts and on the autosampler. The analyte stability in the matrices is verified by analyzing control-quality samples at three concentration levels (low, medium, and high); then after different storage times and at different temperatures (each level of concentration is evaluated at least three times). In fact, analyte stability throughout the validation stage is a requirement for detection and quantification. This means that the integrity of the chemical is ensured to be preserved throughout the analysis procedure. During the last steps of method development, additional stability tests might be performed (Braggio et al., 1996; Viswanathan et al., 2007).
Dilution integrity and matrix effects
In practice, biological samples are not fully compatible with analytical equipment based on their suitability for analysis. In order to perform clinical and pharmacological survey and control of patients, the analysis screening of their physiological liquids and biomarkers is crucial. In bioanalytical techniques, numerous biological matrices might be encountered and each biological matrix presents a different challenge to the analyst as it may contain components that can influence or interfere with the method (Van Eeckhaut et al., 2009). To run across this problem and eliminate or decrease these matrix effects, different measures can be made, such as SPE, LLE, and PP or micro extraction, which is useful as long as the instrumental sensitivity remains adequate (Heller, 2007). Another approach to reduce matrix effects is the optimization of sample preparation and/or chromatographic parameters (Hernandez et al., 2005; Niessen et al., 2006; Xu et al., 2007), by the use of IS to have an idea of the signal threshold. The use of lower flow-rates, flow division, or the need to resort to standard addition are also described (Van Eeckhaut et al., 2009). In most cases, matrix effects are directly related to insufficient purification of the sample under study. During analysis, the effects of the matrix can be reduced by injecting smaller volumes or diluting the sample. However, these solutions will clearly influence the sensitivity of the method and are therefore often inappropriate (Antignac et al., 2005). Nowadays, LC-MS and LC-MS/MS have become a powerful analytical tool. These techniques are sensitive, specific, and allow the analysis of traces in complex mixtures. In addition, LC-MS and LC-MS/MS have been increasingly used in routine clinical laboratories during the last two decades, and their characteristics including specificity, sensitivity, and multianalyte potential make it an ideal alternative to immunoassays and ligand binding assay or HPLC to reduce matrix effect (Nicolas et al., 2004).
The sample dilution procedure should not affect the accuracy or precision of the method. During the validation stage, the analyst is required to prepare a control sample and dilute to the appropriate concentration.
In the process of bioanalytical method development, the accuracy and precision around the mean value may not exceed 15%. Except for the LOQ, the coefficient of variation (CV) should also not be greater than 20%. In a few cases, a dilution of the samples is recommended, and in this case the dilution should be carried out using the same matrix as the sample, but not necessarily from the same subject.Additionally, if the dilution factor varies or if the samples in the study are at levels higher than the dilution quality control (QC), a redilution would be required.
Mobile phase effect
The impact of tiny variations in the solvent ratio of the mobile phase (i.e., <2% of the amount of each component) and the pH buffer, if any, should be discussed and documented for bioanalytical procedures such as chromatographic separation (Polson et al., 2003). On the other hand, GC analysis and additional factors, including slight variations in oven temperature and gas flow rate, must be considered. Evaluation of the following criteria should demonstrate whether the technique can maintain critical separation in the face of expected invariability from column to column and the mobile phase daily variation (Van Eeckhaut et al., 2009).
VALIDATION PARAMETERS
Validation of bioanalytical methods is an experimental protocol applied to ensure that the bioanalytical performance parameters are suitable for the purpose use. More consistency in validation practice is applied for chromatographic methods used in clinical studies (Dadgar and Burnett, 1995; Lindner and Wainer, 1998; Shah et al., 1992, 2000). In general, for chromatographic methods used in biomedical studies, validation procedures in relation to linearity, accuracy, recovery, precision, and LOQ are required. Supplementary parameters that can be tested include the detection limit and the IS and application method. In this section, a correlation between the parameters of validation that must be examined, as well as their acceptance criteria that must be validated are listed in Table 3.
Calibration curve and linearity
The calibration curve and its linearity are defined as the predicted concentration in the samples, and for chromatographic assays, it is the correlation between the compound quantity in the sample and the relative detector response (ICH Q8, 2005). In chromatographic assay, to establish the linear correlation with concentration as a calibration standard, peak area can be used as a response function. The selected calibration model should correctly describe the relationship between the response function and the analyte concentration. The relationship between the measured y-values and the adjusted or residual y-value should be calculated using at least five to eight values (excluding blank values) from the predicted range of concentrations. For each range point made on the same day and at different days, from the equation connecting answer and added concentration (Cr added), a concentration is recalculated (Cr calculated) as well as the corresponding relative error (ER% = [(Cradded − Crcalculated)/Cradded] * 100). For each concentration added, a concentration recalculated average is determined as well as the coefficient of corresponding variation. This coefficient must be below 15%, but 20% of the threshold of quantification remains acceptable (FDA, 2001; Shabir, 2003). Despite the fact that some bioanalysis may involve the application of a nonlinear calibration, it is usual to apply a linear model, based on the principle of parameters estimation using the least squares method. In this approach, the concentration is an independent variable, and the response serves as the dependent variable. The calculation procedure implicitly assumes that the measurement error is the same and is distributed normally for each sample (Hartmann et al., 1998). The linearity of the bioanalytical analysis must be demonstrated as follows: the slope of the linear calibration curve differs from 0 in a statistically significant way, the intercept is not statistically different from 0, and the regression coefficient is not significantly different from 1. If a significant nonzero intercept is obtained, it must be highlighted that there is no effect on the method accuracy (Araujo, 2009; Bischoff et al., 2007).
Recovery
The response of a process spiked matrix standard is calculated as a percentage of the response of a pure standard that has not been pretreated on the sample to estimate the recovery of a bioanalytical technique. It indicates that the method gives a response for the total analyte concentration in the sample (Karnes et al., 1991). It can be proven more clearly by evaluating the outcomes of extracted samples at low, medium, and high enriched matrix concentrations with nonextracted standards that reflect 100% recovery in replicates of at least six (Buick et al., 1990; Lang and Bolton, 1991). In addition, if an IS is used, its recovery should be measured at the method’s concentration level. Although analyte recovery does not have to be 100%, the level of analyte and IS recovery should be accurate, precise, and reproducible. It should also be given (Gao et al., 2011; Lang and Bolton, 1991) by absolute recovery (Sonawane et al., 2014).
Quantification and detection limit
The LOD is the lowest amount of analyte that can be detected but not quantified (Singh et al., 2008). The LOD estimate is vulnerable to error since some bioanalytical laboratories simply measure the lowest quantity of a reference solution that can be detected while others assess the lowest concentration that can be found in the biological sample. The LOQ for individual analytical methods is the smallest amount of analyte in a sample that can be quantitatively determined with adequate precision and accuracy (Murugan et al., 2013b; Sonawane et al., 2014). On the other hand, for validation of the bioanalytical method, the LOQ can estimated by using the relation LOQ = 10 σ/S, where σ represents the response’s standard deviation and S is the calibration curve’s slope. σ is calculated using the intercept, residual standard deviation, regression line, and standard deviation of the blank response (Araujo, 2009). The most commonly applied approaches for estimating LOD are basically the same as those described for the LOQ except for the approach using precision data, which cannot be used here for obvious reasons. In fact, it should be noted that all these approaches only evaluate the pure response of the analytes (Hartmann et al., 1998; Lang and Bolton, 1991; Shah et al., 2000).
 | Table 3. Parameters of bioanalytical methods validation (adapted from Bischoff et al., 2007; FDA, 2001). [Click here to view] |
A simple visual analysis could be adequate for a noninstrumental method. With regard to chromatography methods which have a constant background noise, it can be estimated according to the signal-to-noise ratio.
In such conditions, the LOD will be determined by the concentration at which the signal-to-noise ratio of response equals 3. While, for spectrophotometric techniques, LOD is calculated using the relation 3.3 σ/S, where σ is the standard deviation of the response and S is the slope of the calibration curve. The standard deviation of the response can be obtained. For LOD, an S/N or k-factor equal to or greater than 3 is generally selected either by calculating the residual standard deviation of the regression line or by measuring the standard deviation of the blank response or by computing the standard deviation of the intercept of the Y/x regression line, which is the estimate standard error (Walfish, 2006).
Precision
The precision of a bioanalytical method is defined as the expression of the degree of dispersion between a series of measurements obtained from a multiple samples of the same homogeneous sample under the specified conditions (ICH Q8, 2005). The agreement between replicate measurements of the same sample provides the precision, which is a measure of the random error. Its concept is the replicate values’ relative standard deviation (RSD) or percentage CV (CV%) (Peters et al., 2007).
Precision may be regarded as having a batch component in the assay or repeatability which is described as the ability to replicate the same operator; this is frequently referred to as intra-assay precision: employing the same tools and materials rapidly. In practical application, the ability to repeat the same methodology under different conditions is examined, for example, change of analyst, reagents, or equipment. On subsequent occasions, for example across several weeks or months, it is covered by batch precision and reproducibility, also known as interassay precision. For the validation of new bioanalysis methods for routine use in clinical studies, it is suggested that precision is evaluated at four unique concentrations with six replicates, on four separate steps. This procedure generates data for individual analytes to be analyzed using one-way analysis of variance, which determines the method’s intra-assay and interassay precision at each concentration. To be acceptable, both measures should be within ±20% at each concentration (Shah et al., 1992, 2000).
Accuracy
The accuracy of a method is defined as the closeness of agreement between the test result and the accepted standard value (ICH Q8, 2005). It is obtained by determining the percentage difference (bias %) between the average calculated concentrations and the corresponding nominal concentrations. The accuracy of a bioanalytical method is a measurement of the systematic error or bias and is expressed as the agreement between the measured value and the real value. Accuracy is best reported as percentage bias which is calculated as follows (Wahlich and Carr, 1990):
For examining the correlation between the measured and nominal concentration of the analytes in the spiked substance matrix samples, the accuracy of a bioanalytical method is subsequently established at any concentration. During the certification of a novel bioanalytical technique, the estimated precision will be obtained from the measured concentrations, i.e., from four unique concentrations with six replicates, on four separate steps (4 × 6 × 4 experiments). All findings, excluding those that might be rejected for analytical reasons, including chromatography methods, and the accuracy of the method must be ±15% at each concentration (Deming et al., 1988; Karnes et al., 1991).
Internal standard
During the validation and routine use, the IS is critical in bioanalysis to improve precision and accuracy.
Before sample clean - up, the IS combined with the sample in a predetermined amount, exposing it to the same conditions as the analyte before sample preparation/extraction to allow for losses and errors introduced during the process (Shah et al., 2000). The correct IS should have similar chemical properties to match those of the analyte of interest. In LC-UV and LC-MS analysis, the compound of interest and the corresponding IS provide the ideal situations (Xu et al., 2007). In the same step of sample preparation protocol, a precise quantity of a known IS is added to the sample in order to pursue any procedural loss of the sample which will be accompanied by an equivalent loss of IS. The ratio of the detector response (peak height or area) for the drug and the IS is then used in the calibration and quantification. Furthermore, the calibration curve is created using the ratio between the analyte peak area at each calibration level and the IS peak area at the same range. For these reasons, scientists prefer to use an IS that is structurally similar to the measured drug. One drug may be used as an IS as long as this drug is not a part of the patient’s therapeutic treatment. Guidelines are available for the correct application of the IS in the determination of drugs in biological samples (Stokvis et al., 2005).
Robustness
Following the ICH guidance, the robustness of an analytical method is a measurement of its ability to remain stable through making minor adjustments to the input technique parameters and gives an indication of its reliability in daily use (McPolin, 2009; Murugan et al., 2013a; Sonawane et al., 2014). Robustness may also be described as the capacity to reproduce the technique in multiple laboratories or under various operating conditions without unexpected variations in the result(s) obtained, and a robustness test as a practical tool for evaluating the robustness of bioanalysis.
Carryover
The carryover is a serious problem that may affect the accuracy and precision of a bioanalytical technique. It is more significant in bioanalytical methods using LC-MS-MS, where the dynamic range is very large and therefore affects the reduced values of accuracy and precision. The carryover is mostly induced by the presence of residual analyte of the sample with a high concentration previously tested during the assay. It not only affects the following sample in the series, though depending on the concentration of the prior sample, but may also have an impact on many following samples. Carryover can also be random, as late elution residues from the chromatographic column may affect samples later in the analysis (Hughes et al., 2007). Despite the fact that carryover tests are crucial for method validation, they are not mentioned in the FDA or Agência Nacional de Vigilância Sanitária (ANVISA) guidelines. However, the EMA recommends that carryover should be evaluated by testing series of blank samples after an injection of LOQ. Although acceptance criteria are not included in the EMA guidelines, carryover is generally considered not significant if the area of the blank sample is less than 20% of the analyte peak area in LOQ samples and 5% of the average peak area of the IS in calibration curve standards. If significant carryover is observed, blanks should be injected (the level of carryover affects the number of blanks) during analysis of the study sample for appropriate concentration value estimation. Some analytes tend to adhere to the metallic or polymeric components of the system and may be very difficult to remove. In some cases, an autosampler of a different design may provide injections without transfer effects. However, transfer also depends on the maintenance status of the autosampler and its history of use (Selinger et al., 2014).
Acceptance criteria
The common approach employed runs acceptance criteria as described in the following. For the calibration curve, at minimum six calibration standards making up at minimum 75% of the total number of measurement standards should be within 15% of the nominal value. In the case of LOQ, the difference can be 20%. This requires that if eight calibration values are extracted, at least six (75%) should be used to develop the calibration curve. If nine standards are extracted, at least seven (78%) must be suitable for the calibration curve to be acceptable (Selinger et al., 2014). QC samples are the absolute tool for accepting or rejecting a batch of samples. The “4-6-15” rule is widely accepted, which indicates that six QCs at three concentrations levels in duplicate should be extracted with a batch of test samples (<100), four of these six should be within 15% of the nominal value, and each QCs level must be reflected in these acceptable QCs. In some cases, supplementary acceptance criteria are also included. These, for example, may be as follows:
- The coefficient of determination (r2) of the calibration curve at least 0.99.
- Absence of interference in drug-free samples.
- Concordance of the absolute maximum area or height of an IS.
- Specific QCs, such as hydrolysis QCs, of the test involved.
- Using a dilution QC.
Template validation
On the basis of the validation criteria previously mentioned, it is necessary to follow a sequence of experimental approaches, to take into account all criteria performance appropriately, and to document during full validation and specific acceptance that the method requirements are fulfilled. The evaluation of these parameters allows the exact statement of the analytical procedure as it should be applied in the validation procedure. In order to develop a valid template for the validation of other bioanalytical methods in this field, the following sequence of experiments is required are detailed in Table 4.
New approaches
Before routine bioanalysis, validation of each bioanalytical method in all analytical laboratories is a required step. However, there are many approaches assisting the analyst to suitably conclude that a technique could that an approach, such as accuracy profile, can be determined to be valid.
The concept of total measurement error serves as the basis for the accuracy profile approach, i.e., the combined systematic error measured by bias and random error measured by RSD (Hubert et al., 2003, 2004, 2007). The criteria for the total error are the accuracy of the result. What is required is to provide confidence following the validation process that each of the routine test results the laboratory will achieve in the future will be sufficiently precise. Therefore, to achieve this aim, rather than a complete set of statistical tests, one statistical decision process is used by the accuracy profile approach, i.e., a waiting acceptance range determined using the validation standards’ concentration levels. This range indicates an area where, mainly, a defined fraction of the population of results is expected to be found (i.e., β expectation limits). In this regard, an accuracy profile can be created by using different concentration levels (Hubert et al., 2004, 2007; Rozet et al., 2007). Then this profile is provided to the “a priori” acceptance limits, which are set at ±5% or 15%. This value is a standard limit used in the framework assessment of active substances in pharmaceutical product formulations.
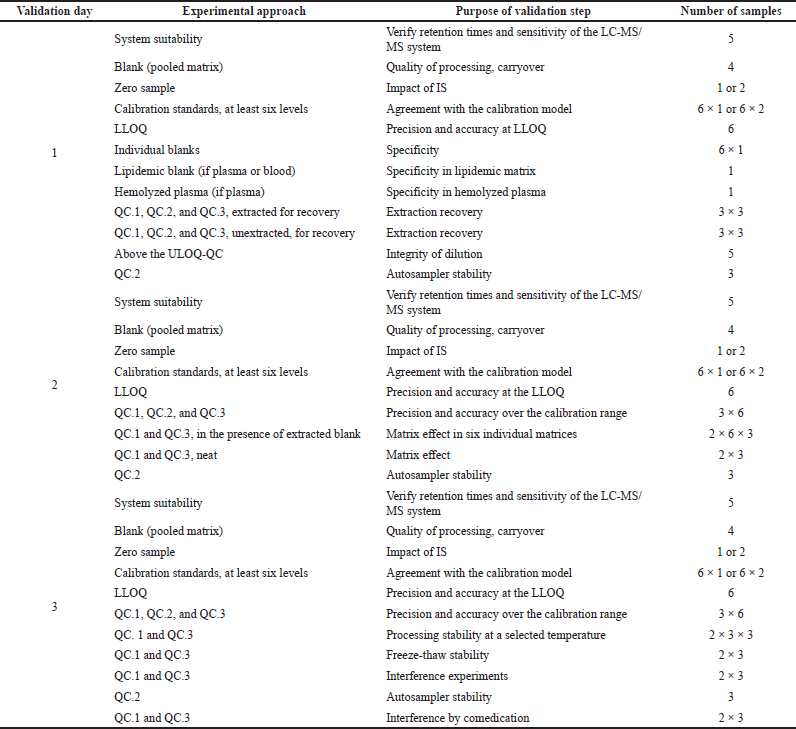 | Table 4. Method validation template. [Click here to view] |
APPLICATION OF THE BIOANALYTICAL METHOD IN THE ROUTINE ANALYSIS OF DRUGS
During the routine development of bioanalytical methods, often attempts have been made to find suitable separation and ionization efficiencies for the analytes. To improve LC-MS techniques, scientists often employ stationary phase selection, mobile phase strength, and available ionization sources. Furthermore, experimental variables such as solvent composition and extraction procedures can affect chromatographic performance but also affect the ionization efficiency of analytes when chromatographic techniques are switched to an ionization source before detection by MS (Crepier, 2018). In general, there are many case studies has been included in Table 5 which used to measure drugs and their metabolites in biological matrix for TDM to optimize therapy of critical dose drugs with a short therapeutic range when there is a high risk of both drug overdosing and underdosing. This table describes case studies of bioanalytical assays validation using various preparation samples and different column separation.
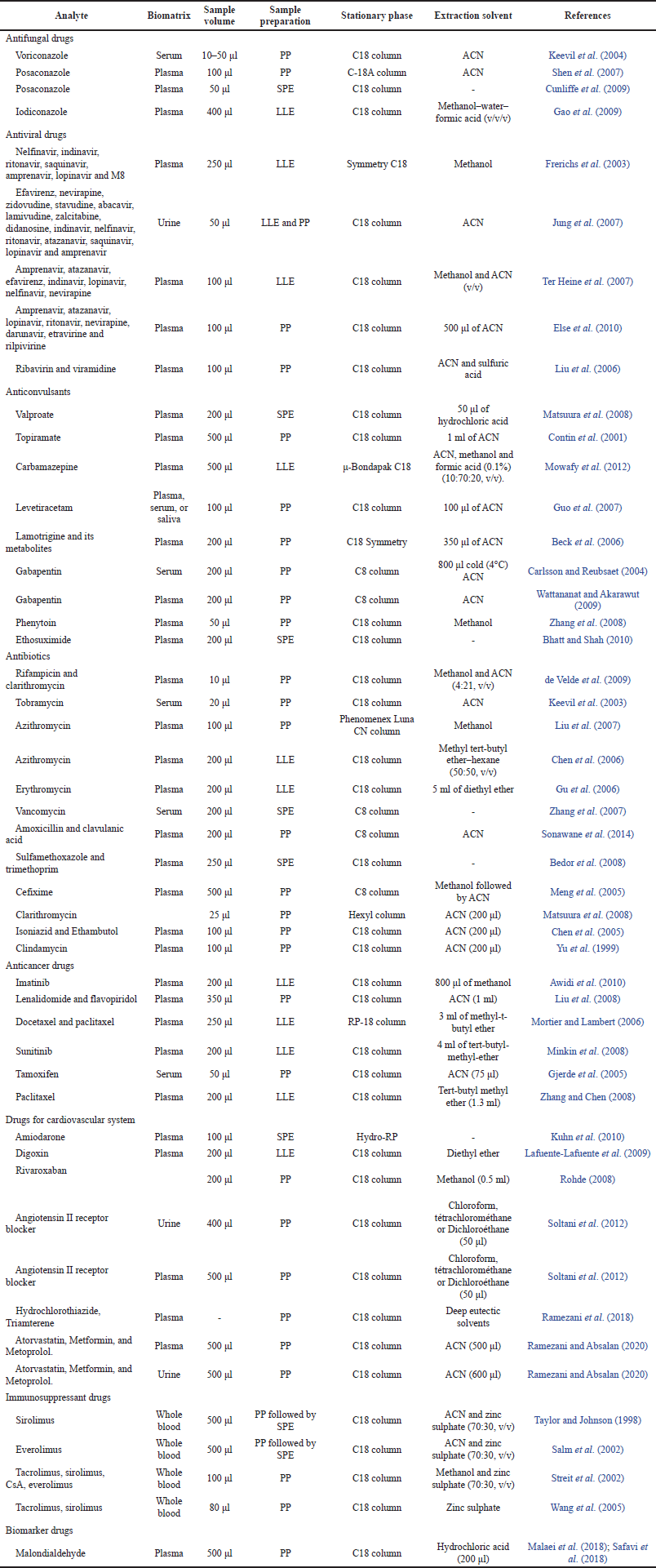 | Table 5. Some case studies methods for drugs analysis TDM. [Click here to view] |
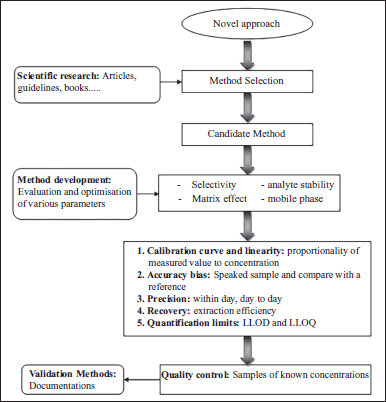 | Figure 3. Various stages for development and validation of bioanalytical method. [Click here to view] |
CYCLE VALIDATION
Validation of a bioanalytical method includes a succession of procedures to demonstrate that the method used makes it possible to quantify an analyte in particular matrices for a precise application. Different parameters define the acceptance and validation of bioanalytical method such as accuracy, recovery, selectivity, specificity, repeatability, linearity, the robustness, and finally the stability of the analyte during the different stages of pretreatment and in the matrix (Causon, 1997; Nicolas et al., 2004; Shah et al., 2000). Validation is a dynamic and adaptable event according to its application and operating conditions. The life cycle of bioanalytical method validation represented in Figure 3 shows that bioanalysis are often described as fixed procedure. This is somewhat the impression given by manuals and other technical standards collections (Feinberg, 2009). However, since any production process, bioanalytical methods are born, evolve, and die. To thoroughly comprehend the significance and importance of validation in the life of bioanalytical technique, it is interesting to describe its life cycle from the moment it is selected until the moment it is abandoned. Finally, a simplified summary of the steps such as full methods validation may cover the experimental plans, which should ideally be applied to the validation of each bioanalytical method.
CONCLUSION
Overall, this review describes a simple practical guide to the validation of bioanalytical methods used in the research and measurement of drugs and their metabolites for TDM. It also pointed out the critical aspects of methodological development according to the international guidelines. For this reason and the need to meet the regulatory requirements of international standards, it has covered and discussed the essential performance characteristics of the validation procedure for bioanalytical methods. On the other hand, we provide guidance to biomedical laboratory staff and simple approaches to use with a scientific background in order to improve the bioanalytical validation process. Today, the commercial availability of a large number of automated rapid tests may reduce the effort involved in the laborious development and validation of LC- MS/MS methods. If all of the objectives are met, LC-MS-MS will be the most robust and widely used technique for the measurement compounds in clinical studies. Finally, other instruments, such as spectroscopic techniques, are developed for the diagnosis of human diseases from their biological samples which can reduce time and cost. This will improve the treatment rate of patients and prevent adverse clinical result.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest regarding the publication of this review.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ackermann BL, Berna MJ, Murphy AT. Recent advances in use of LC/MS/MS for quantitative high-throughput bioanalytical support of drug discovery. Curr Topics Med Chem, 2002; 2(1):53–66. CrossRef
Ali I, Gupta VK, Aboul-Enein HY, Hussain A. Hyphenation in sample preparation: advancement from the micro to the nano world. J Sep Sci, 2008; 31(11):2040–53. CrossRef
Ananthakrishnan N. National workshops on good clinical practices and regulatory requirements for clinical trails in India, training manual by Dr. Urmila Thatte and Dr. Renuka Kulkarni. Indian J Pharmacol, 2005; 37:417.
Antignac JP, de Wasch K, Monteau F, De Brabander H, Andre F, Le Bizec B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal Chim Acta, 2005; 529(1–2):129–36. CrossRef
ANVISA. Resolution RDC 27, minimum requirements for bioanalytical method validation used in studies with the purpose of registration and post-registration of medicines, Brazil. ANVISA, 2012.
ANVISA. Resolution RE no. 899, 23 May 2003. Brazilian Sanitary Surveillance Agency, Brazil, 2003.
Araujo P. Key aspects of analytical method validation and linearity evaluation. J Chromatogr B, 2009; 877(23):2224–34. CrossRef
Ashri NY, Abdel-Rehim M. Sample treatment based on extraction techniques in biological matrices. Bioanalysis, 2011; 3(17):2003–18. CrossRef
Awidi A, Salem II, Najib N, Mefleh R, Tarawneh B. Determination of imatinib plasma levels in patients with chronic myeloid leukemia by high performance liquid chromatography–ultraviolet detection and liquid chromatography–tandem mass spectrometry: methods’ comparison. Leukemia Res, 2010; 34(6):714–7. CrossRef
Bansal SK, Layloff T, Bush ED, Hamilton M, Hankinson EA, Landy JS, Lowes S, Nasr MM, St. Jean PA, Shah VP. Qualification of analytical instruments for use in the pharmaceutical industry: a scientific approach. AAPS Pharmscitech, 2004; 5(1):151–8. CrossRef
Beck O, Öhman I, Nordgren HK. Determination of lamotrigine and its metabolites in human plasma by liquid chromatography-mass spectrometry. Ther Drug Monit, 2006; 28(5):603–7. CrossRef
Bedor DC, Goncalves TM, Ferreira ML, De Sousa CE, Menezes AL, Oliveira EJ, De Santana DP. Simultaneous determination of sulfamethoxazole and trimethoprim in biological fluids for high-throughput analysis: comparison of HPLC with ultraviolet and tandem mass spectrometric detection. J Chromatogr B, 2008; 863(1):46–54. CrossRef
Bhatt M, Shah S. Development of a high-throughput method for the determination of ethosuximide in human plasma by liquid chromatography mass spectrometry. J Chromatogr B, 2010; 878(19):1605–10. CrossRef
Bischoff R, Hopfgartner G, Karnes HT, Lloyd HT, Phillips TM, Tsikas D, Xu G. Summary of a recent workshop/conference report on validation and implementation of bioanalytical methods: implications on manuscript review in the Journal of Chromatography B. J Chromatogr B, 2007; 860:1–3. CrossRef
Blanchard J, Boyle JO, Van Wagenen S. Determination of the partition coefficients, acid dissociation constants, and intrinsic solubility of carbenoxolone. J Pharm Sci, 1988; 77(6):548–52. CrossRef
Boulanger B, Chiap P, Dewé W, Crommen J. An analysis of the SFSTP guide on validation of chromatographic bioanalytical methods: progresses and limitations. J Pharm Biomed Anal, 2003; 32(4–5):753–65. CrossRef
Braggio S, Barnaby RJ, Grossi P, Cugola M. A strategy for validation of bioanalytical methods. J Pharm Biomed Anal, 1996; 14(4):375–88. CrossRef
Buick AR, Doig MV, Jeal SC, Land GS, McDowall RD. Method validation in the bioanalytical laboratory. J Pharm Biomed Anal, 1990; 8(8–12):629–37. CrossRef
Burgess RR. Protein precipitation techniques. Methods in enzymology. Elsevier, vol. 463, pp 331–42, 2009. CrossRef
Capiau S, Veenhof H, Koster RA, Bergqvist Y, Boettcher M, Halmingh O, Keevil BG, Koch BC, Linden R, Pistos C, Stolk LM. Official international association for therapeutic drug monitoring and clinical toxicology guideline: development and validation of dried blood spot–based methods for therapeutic drug monitoring. Ther Drug Monit, 2019; 41(4):409–30. CrossRef
Carlsson K, Reubsaet J. Sample preparation and determination of gabapentin in venous and capillary blood using liquid chromatography–tandem mass spectrometry. J Pharm Biomed Anal, 2004; 34(2):415–23. CrossRef
Causon R. Validation of chromatographic methods in biomedical analysis viewpoint and discussion. J Chromatogr B Biomed Sci Appl, 1997; 689(1):175–80. CrossRef
Chang MA, Ji Q, Zhang J, El-Shourbagy TA. Historical review of sample preparation for chromatographic bioanalysis: pros and cons. Drug Dev Res, 2007; 68(3):107–33. CrossRef
Chen BM, Liang YZ, Chen X, Liu SG, Deng FL, Zhou P. Quantitative determination of azithromycin in human plasma by liquid chromatography–mass spectrometry and its application in a bioequivalence study. J Pharm Biomed Anal, 2006; 42(4):480–7. CrossRef
Chen X, Song B, Jiang H, Yu K, Zhong D. A liquid chromatography/tandem mass spectrometry method for the simultaneous quantification of isoniazid and ethambutol in human plasma. Rapid Commun Mass Spectrom, 2005; 19(18):2591–6. CrossRef
Contin M, Riva R, Albani F, Baruzzi A. Simple and rapid liquid chromatographic-turbo ion spray mass spectrometric determination of topiramate in human plasma. J Chromatogr B Biomed Sci Appl, 2001; 761(1):133–7. CrossRef
Cremers S, Guha N, Shine B. Therapeutic drug monitoring in the era of precision medicine: opportunities! Br J Clin Pharmacol, 2016; 82(4):900–2. CrossRef
Crepier J. La chromatographie en phase supercritique couplée avec une détection UV et spectrométrie de masse haute résolution pour l’analyse d’échantillons complexes: application aux bio-huiles. University of Lyon, Lyon, France, 2018.
Cunliffe JM, Noren CF, Hayes RN, Clement RP, Shen JX. A high-throughput LC-MS/MS method for the quantitation of posaconazole in human plasma: implementing fused core silica liquid chromatography. J Pharm Biomed Anal, 2009; 50(1):46–52. CrossRef
Dadgar D, Burnett PE. Issues in evaluation of bioanalytical method selectivity and drug stability. J Pharm Biomed Anal, 1995; 14(1–2):23–31. CrossRef
Deming S, Michotte Y, Massart DL, Kaufman L, Vandeginste B. Chemometrics: a textbook. Elsevier, 1988.
de Velde F, Alffenaar JW, Wessels AM, Greijdanus B, Uges DR. Simultaneous determination of clarithromycin, rifampicin and their main metabolites in human plasma by liquid chromatography–tandem mass spectrometry. J Chromatogr B, 2009; 877(18–19):1771–7. CrossRef
Dunér K, Bäckström J, Magnell N, Svennberg H, Ahnoff M, Logren U. Determination of ximelagatran, melagatran and two intermediary metabolites in plasma by mixed-mode solid phase extraction and LC–MS/MS. J Chromatogr B, 2007; 852(1–2):317–24. CrossRef
Else L, Watson V, Tjia J, Hughes A, Siccardi M, Khoo S, Back D. Validation of a rapid and sensitive high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) assay for the simultaneous determination of existing and new antiretroviral compounds. J Chromatogr B, 2010; 878(19):1455–65. CrossRef
Ermer J, Miller JHM. (Eds.). Method validation in pharmaceutical analysis: a guide to best practice. John Wiley & Sons, 2006. CrossRef
Feinberg M. Labo-Stat: guide de validation des méthodes d’analyse. Éd. Tec & doc, Paris, France, 2009.
FDA. Guidance for industry: bioanalytical method validation. 2001. Available via http://www.fda.gov/cder/Guidance/4252fnl.pdf
FDA Thailand. Guidelines for the conduct of bioavailability and bioequivalence studies. Drug Control Division, Ministry of Public Health, Bangkok, Thailand, 2001.
Food and Drug Administration. Guidance for industry. Center for Drug Evaluation and Research (CDER), 2011.
Food and Drug Administration. Guidance for industry: bioanalytical method validation. FDA, pp 1–22, 2016.
Food and Drug Administration of the United States. Guidance for industry—bioanalytical method validation. US Department of Health and Human Services, Center for Drug Evaluation and Research and Center for Veterinary Medicine, 2003.
Frerichs VA, DiFrancesco R, Morse GD. Determination of protease inhibitors using liquid chromatography–tandem mass spectrometry. J Chromatogr B, 2003; 787(2):393–403. CrossRef
Gao L, Li J, Kasserra C, Song Q, Arjomand A, Hesk D, Chowdhury SK. Precision and accuracy in the quantitative analysis of biological samples by accelerator mass spectrometry: application in microdose absolute bioavailability studies. Anal Chem, 2011; 83(14):5607–16. CrossRef
Gao S, Tao X, Sun L, Sheng C, Zhang W, Yun Y, Li J, Miao H, Chen W. An liquid chromatography-tandem mass spectrometry assay for determination of trace amount of new antifungal drug iodiconazole in human plasma. J Chromatogr B, 2009; 877(4):382–6. CrossRef
Gjerde J, Kisanga ER, Hauglid M, Holm PI, Mellgren G, Lien EA. Identification and quantification of tamoxifen and four metabolites in serum by liquid chromatography-tandem mass spectrometry. J Chromatogr A, 2005; 1082(1):6–14. CrossRef
Gu Y, Wang G, Sun J. Simultaneous determination of erythromycin ethylsuccinate and its metabolite erythromycin in human plasma using liquid chromatography–electrospray ionization mass spectrometry for clinical study. J Pharm Biomed Anal, 2006; 40(3):737–43. CrossRef
Guo T, Oswald LM, Mendu DR, Soldin SJ. Determination of levetiracetam in human plasma/serum/saliva by liquid chromatography-electrospray tandem mass spectrometry. Clin Chim Acta, 2007; 375(1–2):115–8. CrossRef
Hartmann C, Smeyers-Verbeke J, Massart DL, McDowall RD. Validation of bioanalytical chromatographic methods. J Pharm Biomed Anal, 1998; 17(2):193–218. CrossRef
Heller DN. Ruggedness testing of quantitative atmospheric pressure ionization mass spectrometry methods: the effect of co-injected matrix on matrix effects. Rapid Commun Mass Spectrom, 2007; 21(5):644–52. CrossRef
Hernandez F, Sancho J, Pozo O. Critical review of the application of liquid chromatography/mass spectrometry to the determination of pesticide residues in biological samples. Anal Bioanal Chem, 2005; 382(4):934–46. CrossRef
Horie H, Kidowaki T, Koyama Y, Endo T, Homma K, Kambegawa A, Aoki N. Specificity assessment of immunoassay kits for determination of urinary free cortisol concentrations. Clin Chim Acta, 2007; 378(1–2):66–70. CrossRef
Hsieh Y, Brisson J, Wang G. Fast HPLC-MS/MS for small molecules. Am Pharm Rev, 2003; 6:14–20.
Hubert P, Nguyen-Huu JJ, Boulanger B, Chapuzet E, Cohen N, Compagnon PA, Dewé W, Feinberg M, Laurentie M, Mercier N, Muzard G, Valat L, Rozet E. Harmonization of strategies for the validation of quantitative analytical procedures: a SFSTP proposal—part I. J Pharm Biomed Anal, 2004; 36(3):579–86. CrossRef
Hubert P, Nguyen-Huu JJ, Boulanger B, Chapuzet E, Cohen N, Compagnon PA, Dewé W, Feinberg M, Laurentie M, Mercier N, Muzard G, Valat L, Rozet E. Harmonization of strategies for the validation of quantitative analytical procedures: a SFSTP proposal–part II. J Pharm Biomed Anal, 2007; 45(1):70–81. CrossRef
Hubert P, Nguyen-Huu J, Laurentie M, Mercier N, Muzard G. Validation Des procédures analytiques quantitatives harmonisation des démarches. STP Pharm Pratiques, 2003; 13(3):101–38.
Hughes NC, Wong EY, Fan J, Bajaj N. Determination of carryover and contamination for mass spectrometry-based chromatographic assays. AAPS J, 2007; 9(3):E353–60. CrossRef
ICH Guideline. Bioanalytical method validation M10. ICH, 2019. CrossRef
ICH Q8. International Conference on Harmonization (ICH) of technical requirements for registration of pharmaceuticals for human use topic Q9: quality risk management. ICH, Geneva, Switzerland, 2005.
Jung BH, Rezk NL, Bridges AS, Corbett AH, Kashuba AD. Simultaneous determination of 17 antiretroviral drugs in human plasma for quantitative analysis with liquid chromatography–tandem mass spectrometry. Biomed Chromatogr, 2007; 21(10):1095–104. CrossRef
Karnes HT, Shiu G, Shah VP. Validation of bioanalytical methods. Pharm Res, 1991; 8(4):421–6. CrossRef
Keevil BG, Lockhart SJ, Cooper DP. Determination of tobramycin in serum using liquid chromatography–tandem mass spectrometry and comparison with a fluorescence polarisation assay. J Chromatogr B, 2003; 794(2):329–35. CrossRef
Keevil BG, Newman S, Lockhart S, Howard SJ, Moore CB, Denning DW. Validation of an assay for voriconazole in serum samples using liquid chromatography-tandem mass spectrometry. Ther Drug Monit, 2004; 26(6):650–7. CrossRef
KFDA. Guidance document for bioequivalence studies 2008. KFDA, 2011. CrossRef
Kuhn J, Götting C, Kleesiek K. Simultaneous measurement of amiodarone and desethylamiodarone in human plasma and serum by stable isotope dilution liquid chromatography–tandem mass spectrometry assay. J Pharm Biomed Anal, 2010; 51(1):210–6. CrossRef
Lafuente-Lafuente C, Alvarez JC, Leenhardt A, Mouly S, Extramiana F, Caulin C, Funck-Brentano C, Bergmann JF. Amiodarone concentrations in plasma and fat tissue during chronic treatment and related toxicity. Br J Clin Pharmacol, 2009; 67(5):511–9. CrossRef
Lang J, Bolton S. A comprehensive method validation strategy for bioanalytical applications in the pharmaceutical industry—2. Statistical analyses. J Pharm Biomed Anal, 1991; 9(6):435–42. CrossRef
Lazaou K, de Geyter T, de Reu L, Zhao Y, Sandra P. Applicability of a benchtop LC-DAD-MSD for quality control on the pharmaceutical industry. LC GC Europe, 2000; 13:340–56.
Lee JS, Ettinger B, Stanczyk FZ, Vittinghoff E, Hanes V, Cauley JA, Chandler W, Settlage J, Beattie MS, Folkerd E, Dowsett M, Grady D, Cummings SR. Comparison of methods to measure low serum estradiol levels in postmenopausal women. J Clin Endocrinol Metab, 2006; 91(10):3791–7. CrossRef
Lee MS, Kerns EH. LC/MS applications in drug development. Mass Spectrom Rev, 1999; 18(3-4):187–279. CrossRef
Lindner W, Wainer I. Requirements for initial assay validation and publication in J. Chromatography B. J Chromatogr B Biomed Sci Appl, 1998; 707(1–2):1–2.
Liu F, Xu Y, Huang J, Gao S, Guo Q. Sensitive liquid chromatography/mass spectrometry assay for the quantification of azithromycin in human plasma. Biomed Chromatogr, 2007; 21(12):1272–8. CrossRef
Liu Q, Farley KL, Johnson AJ, Muthusamy N, Hofmeister CC, Blum KA, Schaaf LJ, Grever MR, Byrd JC, Dalton JT, Phelps MA. Development and validation of a highly sensitive liquid chromatography/mass spectrometry method for simultaneous quantification of lenalidomide and flavopiridol in human plasma. Ther Drug Monit, 2008; 30(5):620. CrossRef
Liu Y, Xu C, Yan R, Lim C, Yeh LT, Lin CC. Sensitive and specific LC–MS/MS method for the simultaneous measurements of viramidine and ribavirin in human plasma. J Chromatogr B, 2006; 832(1):17–23. CrossRef
Malaei R, Ramezani AM, Absalan G. Analysis of malondialdehyde in human plasma samples through derivatization with 2, 4-dinitrophenylhydrazine by ultrasound-assisted dispersive liquid–liquid microextraction-GC-FID approach. J Chromatogr B, 2018; 1089:60–9. CrossRef
Matsuura K, Ohmori T, Nakamura M, Itoh Y, Hirano K. A simple and rapid determination of valproic acid in human plasma using a non-porous silica column and liquid chromatography with tandem mass spectrometric detection. Biomed Chromatogr, 2008; 22(4):387–93. CrossRef
McPolin O. Validation of analytical methods for pharmaceutical analysis. 2009. Available via Lulu.com
Meng F, Chen X, Zeng Y, Zhong D. Sensitive liquid chromatography–tandem mass spectrometry method for the determination of cefixime in human plasma: application to a pharmacokinetic study. J Chromatogr B, 2005; 819(2):277–82. CrossRef
Minkin P, Zhao M, Chen Z, Ouwerkerk J, Gelderblom H, Baker SD. Quantification of sunitinib in human plasma by high-performance liquid chromatography–tandem mass spectrometry. J Chromatogr B, 2008; 874(1–2):84–8. CrossRef
Mortier KA, Lambert WE. Determination of unbound docetaxel and paclitaxel in plasma by ultrafiltration and liquid chromatography–tandem mass spectrometry. J Chromatogr A, 2006; 1108(2):195–201. CrossRef
Mou L, Jiang X. Materials for microfluidic immunoassays: a review. Adv Healthc Mater, 2017; 6(15):1601403. CrossRef
Mowafy HA, Alanazi FK, El Maghraby GM. Development and validation of an HPLC–UV method for the quantification of carbamazepine in rabbit plasma. Saudi Pharm J, 2012; 20(1):29–34. CrossRef
Murugan S, Elayaraja A, Chandrakala K, Ramaiah P, Vulchi C. A review on method development and validation by using HPLC. Int J Novel Trends Pharm Sci, 2013a; (4):78–81.
Murugan S, Pravallika N, Sirisha P, Chandrakala K. A review on bioanalytical method development and validation by using LC-MS/MS. J Chem Pharm Sci, 2013b; 6(1):41–5.
Nicolas O, Farenc C, Bressolle F. Stratégie de validation de méthodes de dosage en bioanalyse en vue d’études pharmacocinétiques et toxicologiques Ann Toxicol Anal, 2004; 6:118–27. CrossRef
Niessen W, Manini P, Andreoli R. Matrix effects in quantitative pesticide analysis using liquid chromatography–mass spectrometry. Mass Spectrom Rev, 2006; 25(6):881–99. CrossRef
Nováková L, Vl?ková H. A review of current trends and advances in modern bio-analytical methods: chromatography and sample preparation. Anal Chim Acta, 2009; 656(1–2):8–35. CrossRef
Ohno Y. Notification from the Ministry of Health, Labour, and Welfare-clinical pharmacokinetics of pharmaceuticals and methods for drug interaction studies. Clin Eval, 2001; 29(1):153–66.
Peters FT, Drummer OH, Musshoff F. Validation of new methods. Forensic Sci Int, 2007; 165(2–3):216–24. CrossRef
Peters FT, Maurer HH. Bioanalytical method validation and its implications for forensic and clinical toxicology—a review. Validation in chemical measurement. Springer, pp 1–9, 2002. CrossRef
Polson C, Sarkar P, Incledon B, Raguvaran V, Grant R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography–tandem mass spectrometry. J Chromatogr B, 2003; 785(2):263–75. CrossRef
Poole CF. New trends in solid-phase extraction. Trends Anal Chem, 2003; 22(6):362–73. CrossRef
Ramezani AM, Absalan G. Employment of a natural deep eutectic solvent as a sustainable mobile phase additive for improving the isolation of four crucial cardiovascular drugs by micellar liquid chromatography. J Pharm Biomed Anal, 2020; 186:113259. CrossRef
Ramezani AM, Absalan G, Ahmadi R. Green-modified micellar liquid chromatography for isocratic isolation of some cardiovascular drugs with different polarities through experimental design approach. Anal Chim Acta, 2018; 1010:76–85. CrossRef
Raynie DE. Modern extraction techniques. Anal Chem, 2006; 78(12):3997–4004. CrossRef
Remane D, Meyer MR, Peters FT, Wissenbach DK, Maurer HH. Fast and simple procedure for liquid–liquid extraction of 136 analytes from different drug classes for development of a liquid chromatographic-tandem mass spectrometric quantification method in human blood plasma. Anal Bioanal Chem, 2010; 397(6):2303–14. CrossRef
Riley CM, Rosanske TW. Development and validation of analytical methods. Elsevier, 1996.
Rohde G. Determination of rivaroxaban–a novel, oral, direct Factor Xa inhibitor–in human plasma by high-performance liquid chromatography–tandem mass spectrometry. J Chromatogr B, 2008; 872(1–2):43–50. CrossRef
Rozet E, Wascotte V, Lecouturier N, Préat V, Dewé W, Boulanger B, Hubert P. Improvement of the decision efficiency of the accuracy profile by means of a desirability function for analytical methods validation: application to a diacetyl-monoxime colorimetric assay used for the determination of urea in transdermal iontophoretic extracts. Anal Chim Acta, 2007; 591(2):239–47. CrossRef
Ryan BJ. Differential precipitation and solubilization of proteins. Methods Mol Biol, 2011; 681:203–13. CrossRef
Safavi A, Ahmadi R, Ramezani AM. Vortex-assisted liquid-liquid microextraction based on hydrophobic deep eutectic solvent for determination of malondialdehyde and formaldehyde by HPLC-UV approach. Microchem J, 2018; 143:166–74. CrossRef
Salm P, Taylor PJ, Lynch SV, Pillans PI. Quantification and stability of everolimus (SDZ RAD) in human blood by high-performance liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr B, 2002; 772(2):283–90. CrossRef
Saudi Food and Drug Authority. Bioequivalence requirements guidelines. Drug Sector, Kingdom of Saudi Arabia, Draft May 2005. 2005.
Selinger K, Fung EN, Bryan P. Bioanalytical method validation and bioanalysis in regulated settings. Specification of drug substances and products. Elsevier, pp 325–63, 2014. CrossRef
Shabir GA. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J Chromatogr A, 2003; 987(1–2):57–66. CrossRef
Shah VP, Bansal S. Historical perspective on the development and evolution of bioanalytical guidance and technology. Bioanalysis, 2011; 3(8):823–7. CrossRef
Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD, Pittman KA. Analytical methods validation: bioavailability, bioequivalence, and pharmacokinetic studies. J Pharm Sci, 1992; 81(3):309–12. CrossRef
Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A. Bioanalytical method validation—a revisit with a decade of progress. Pharm Res, 2000; 17(12):1551–7. CrossRef
Shen JX, Krishna G, Hayes RN. A sensitive liquid chromatography and mass spectrometry method for the determination of posaconazole in human plasma. J Pharm Biomed Anal, 2007; 43(1):228–36. CrossRef
Singh U, Pandey S, Pandey P, Keshri P, Wal P. Bioanalytical method development and validation. Express Pharm, 2008; 2:1–8.
Smith G. European Medicines Agency guideline on bioanalytical method validation: what more is there to say? Bioanalysis, 2012; 4(8):865–8. CrossRef
Soltani S, Ramezani AM, Soltani N, Jouyban A. Analysis of losartan and carvedilol in urine and plasma samples using a dispersive liquid–liquid microextraction isocratic HPLC–UV method. Bioanalysis, 2012; 4(23):2805–21. CrossRef
Sonawane LV, Poul BN, Usnale SV, Waghmare PV, Surwase LH. Bioanalytical method validation and its pharmaceutical application-a review. Pharm Anal Acta, 2014; 5(288):2. CrossRef
Souverain S, Rudaz S, Veuthey JL. Protein precipitation for the analysis of a drug cocktail in plasma by LC–ESI–MS. J Pharm Biomed Anal, 2004; 35(4):913–20. CrossRef
Stokvis E, Rosing H, Beijnen JH. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun Mass Spectrom, 2005; 19(3):401–7. CrossRef
Streit F, Armstrong VW, Oellerich M. Rapid liquid chromatography–tandem mass spectrometry routine method for simultaneous determination of sirolimus, everolimus, tacrolimus, and cyclosporin A in whole blood. Clin Chem, 2002; 48(6):955–8. CrossRef
Taieb J, Benattar C, Birr AS, Lindenbaum A. Limitations of steroid determination by direct immunoassay. Clinical chemistry 2002; 48(3):583–5. CrossRef
Taylor PJ, Johnson AG. Quantitative analysis of sirolimus (Rapamycin) in blood by high-performance liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr B Biomed Sci Appl, 1998; 718(2):251–7. CrossRef
Ter Heine R, Alderden-Los CG, Rosing H, Hillebrand MJ, Van Gorp EC, Huitema AD, Beijnen JH. Fast and simultaneous determination of darunavir and eleven other antiretroviral drugs for therapeutic drug monitoring: method development and validation for the determination of all currently approved HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors in human plasma by liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom, 2007; 21(15):2505–14. CrossRef
Tiller PR, Romanyshyn LA, Neue UD. Fast LC/MS in the analysis of small molecules. Analyt Bioanal Chem, 2003; 377(5):788–802. CrossRef
Tiwari G, Tiwari R. Bioanalytical method validation: an updated review. Pharm Methods, 2010; 1(1):25–38. CrossRef
US-FDA. Bioanalytical method validation, draft guidance. US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research, Rockville, MD, 2018. Available via https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf
US Food and Drug Administration. Guidance for industry: bioanalytical method validation, draft guidance. US Food and Drug Administration, Silver Spring, MD, pp 4–24, 2013.
Valcárcel M, Gómez-Hens A, Rubio S. Selectivity in analytical chemistry revisited. Trends Anal Chem, 2001; 20(8):386–93. CrossRef
van Amsterdam P, Lausecker B, Luedtke S, Timmerman P, Brudny-Kloeppel M. Towards harmonized regulations for bioanalysis: moving forward! Bioanalysis, 2010; 2(4):689–91. CrossRef
Van Eeckhaut A, Lanckmans K, Sarre S, Smolders I, Michotte Y. Validation of bioanalytical LC–MS/MS assays: evaluation of matrix effects. J Chromatogr B, 2009; 877(23):2198–207. CrossRef
van Nuland M, Rosing H, Schellens JH, Beijnen JH. Bioanalytical LC–MS/MS validation of therapeutic drug monitoring assays in oncology. Biomed Chromatogr, 2010; 34(1):e4623. CrossRef
Vessman J. Selectivity or specificity? Validation of analytical methods from the perspective of an analytical chemist in the pharmaceutical industry. J Pharm Biomed Anal, 1996; 14(8–10):867–9. CrossRef
Viswanathan C. Regulatory observations in bioanalytical determinations. Bioanalysis, 2010; 2(7):1325–9. CrossRef
Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, Sailstad J, Shah VP, Skelly JP, Swann PG, Weiner R. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm Res, 2007; 24(10):1962–73. CrossRef
Vogeser M, Kirchhoff F. Progress in automation of LC-MS in laboratory medicine. Clin Biochem, 2011; 44(1):4–13. CrossRef
Wahlich J, Carr G. Chromatographic system suitability tests—what should we be using? J Pharm Biomed Anal, 1990; 8(8–12):619–23. CrossRef
Walfish S. Analytical methods: a statistical perspective on the ICH Q2A and Q2B guidelines for validation of analytical methods. BioPharm Int, 2006; 19(12):1–6.
Wang S, Magill JE, Vicente FB. A fast and simple high-performance liquid chromatography/mass spectrometry method for simultaneous measurement of whole blood tacrolimus and sirolimus. Arch Pathol Lab Med, 2005; 129(5):661–5. CrossRef
Wattananat T, Akarawut W. Validated LC-MS-MS method for the determination of gabapentin in human plasma: application to a bioequivalence study. Oxford University Press, Oxford, UK, 2009. CrossRef
Whitmire M, Ammerman J, De Lisio P, Killmer J, Kyle D, Mainstone E, Porter L, Zhang T. LC-MS/MS bioanalysis method development, validation, and sample analysis: points to consider when conducting nonclinical and clinical studies in accordance with current regulatory guidances. J Anal Bioanal Tech, 2011; 4:2. CrossRef
Xu RN, Fan L, Rieser MJ, El-Shourbagy TA. Recent advances in high-throughput quantitative bioanalysis by LC–MS/MS. J Pharm Biomed Anal, 2007; 44(2):342–55. CrossRef
Yu LL, Chao CK, Liao WJ, Twu TY, Liu CM, Yang TH, Lin ET. Determination of clindamycin in human plasma by liquid chromatography–electrospray tandem mass spectrometry: application to the bioequivalence study of clindamycin. J Chromatogr B Biomed Sci Appl, 1999; 724(2):287–94. CrossRef
Zhang SQ, Chen GH. Determination of paclitaxel in human plasma by UPLC-MS-MS. J Chromatogr Sci, 2008; 46(3):220–4. CrossRef
Zhang T, Watson DG, Azike C, Tettey JN, Stearns AT, Binning AR, Payne CJ. Determination of vancomycin in serum by liquid chromatography–high resolution full scan mass spectrometry. J Chromatogr B, 2007; 857(2):352–6. CrossRef
Zhang Y, Mehrotra N, Budha NR, Christensen ML, Meibohm B. A tandem mass spectrometry assay for the simultaneous determination of acetaminophen, caffeine, phenytoin, ranitidine, and theophylline in small volume pediatric plasma specimens. Clin Chim Acta, 2008; 398(1–2):105–12. CrossRef