
INTRODUCTION
It is estimated that breast cancer is the most common cancer diagnosed in women around the world, overtaking lung cancer as the most common cancer. There were 2.3 million new cases of breast cancer detected in women in 2020, and 685,000 deaths directly attributed to the disease (https://www.who.int). Triple-negative breast cancers (TNBC) account for 10%–15% of invasive breast cancers. These tumors lack estrogen receptors (ER), progesterone receptors (PR), and human epidermal growth factor receptor-2 (HER2). The incidence of these cancers is higher among women who have a mutation in the Breast Cancer gene 1 (https://www.cancer.org). Because TNBC lacks hormone receptors (ER/PR), it cannot benefit from endocrine therapy, so chemotherapy is only the primary treatment option (Gangi et al., 2014). Despite the use of clinically approved drugs such as taxanes and anthracyclines (Engebraaten et al., 2013), systemic chemotherapy results in a poor response, high toxicity, and the development of multidrug resistance (MDR) (Dai et al., 2015; Lord and Ashworth, 2016). Paclitaxel (PTX) is used to treat several types of cancers, including breast, ovarian, and lung cancers, and is a white crystalline solid categorized as a Biopharmaceutics Classification System (BCS) class IV drug. It is poorly soluble in water, and the solubility is pH dependent, with increased solubility at lower pH values. PTX has a relatively high partition coefficient, reflects a high affinity for lipids, can easily cross cell membranes, as well as shows high binding with plasma proteins. Numerous studies have proven that PTX is an effective treatment for metastatic and triple-negative breast cancer. PTX acts by binding to the beta subunit of tubulin, a key protein in microtubules, and stabilizes its structure. This prevents the microtubules from disassembling, leading to cell cycle arrest in the G2/M phase that eventually results in apoptosis. Even though it is effective in treating a variety of solid tumors, including heterogeneous breast cancers, its poor solubility limits its use (Ajani et al., 1994; Kato et al., 2011; Kavallaris, 2010; Liu et al., 2016). Lapatinib (LPB) is a small-molecule tyrosine kinase (TK) inhibitor that blocks both epidermal growth factor receptor (EGFR) and HER2 in the treatment of breast cancer. LPB is a crystalline compound that has poor water solubility and is classified as a BCS class II drug. As a result of its high partition coefficient and strong affinity for lipids, LPB is readily able to cross cell membranes. The LPB is rapidly absorbed from the gastrointestinal tract and has a high affinity for binding to plasma proteins. LPB works by binding to the TK domains of EGFR-1 and EGFR-2 (HER2) and serves as an adenosine triphosphate (ATP) competitor by inhibiting phosphorylation and downstream signaling, required for cell growth and survival, differentiation, angiogenesis, and apoptosis (Medina and Goodin, 2008). Several studies have demonstrated promising results when LPB is used to treat TNBC (Baselga et al., 2005; Dickler et al., 2009), as approximately three-fourths of these tumors express the EGFR, as determined by immunohistochemical analysis (Nielsen et al., 2004).
Breast cancer patients frequently develop MDR and relapse during treatment. Several mechanisms can lead to the development of MDR, but transporter-mediated drug efflux has been identified as the mechanism that has been extensively evaluated. (Wind and Holen, 2011). Several ATP-binding cassette (ABC) transporters have been associated with MDR mechanisms in various types of breast cancer (Sharom, 2008). There was a marked increase in the expression of multidrug-resistant protein-1 (MDR1/ABCC1), breast cancer resistance protein (BCRP/ABCG2), and multidrug-resistant protein-8 (MRP8/ABCC11) in TNBC compared to other subtypes of breast cancer (Xu et al., 2017; Yamada et al., 2013). Acquired resistance to multiple chemotherapeutic drugs (multiple drug resistance), including taxanes, is a key obstacle in the clinical management of triple-negative breast cancer. Resistance to PTX in breast tumor cells is often acquired by active drug ejection due to elevated MDR-1 or/and BCRP activity (Fletcher et al., 2016; Furstova et al., 2016). MDR is a critical issue that must be overcome in order to maximize the clinical benefit of present chemotherapeutic and targeted therapies in TNBC.
By targeting key pathways synergistically or additively, combination therapy enhances efficacy compared to monotherapy for cancer. However, due to the fact that each drug has its own distinct pharmacokinetic profile, it is exceedingly difficult to deliver dual or multi-component therapies concurrently to the tumor site in the optimal proportion by integrating existing preparations (Mi et al., 2018; Zacharoula et al., 2018). Nonetheless, nanoformulations can be employed to deliver several drugs to a distinct site at once or in a precise order. As nanoparticles are largely taken up by cells by endocytosis and help in the evacuation of different payloads from the cell cytoplasm prior to release, the mechanism of drug efflux driven by ABC cassette transporters is naturally reduced when nanoformulations are employed as transport carriers (Cruz et al., 2011, 2014). Several studies have shown that liposomes (Lps), among other nanoparticulate systems, facilitate the delivery of anticancer drugs into the nucleus and allow them to remain active for a longer period. The application of Lps can provide a wealth of advantages, including the protection of drugs against degradation, the optimization of their pharmacokinetic properties, and the reduction of their lethality when delivered to healthy tissue (Akbarzadeh et al., 2013). Despite multiple studies demonstrating the co-delivery of PTX and LPB (Cruz et al., 2017; Hu et al., 2015; Levit et al., 2020), using multi-component carriage systems for various cancer applications, this combination has shown promising results for ovarian cancers in recent years (Vergara et al., 2012). However, it is crucial to emphasize that the management of MDR1-mediated triple-negative breast tumors with the combination of PTX and LPB has not been studied. In light of these data, we designed a dual-loaded nano-scale formulation that encompasses both PTX and LPB in an effort to circumvent MDR in triple-negative breast cancer models with synergistic effectiveness.
To improve site-targeted distribution and anti-cancer impacts, we thus present the fabrication of Pluronic® P123-coated PTX/LPB dual-loaded Lps, which use the nanoscale benefits of polymer. Pluronic® block copolymers include hydrophobic (hydrophobic) polypropylene oxide (PPO) and hydrophilic polyethylene oxide (PEO) moieties, respectively, which inhibit macrophage capture in-vitro and in-vivo and prolong blood circulation (Anirudhan et al., 2021; Illum and Davis, 1984; Lin et al., 2018; Redhead et al., 2001). We determined the size by dimension, shape, zeta potential, drug loading, and entrapment efficiency of PTX and LPB dual-loaded Lps, as well as the rate at which the drug(s) was released from the fabricated Lps in vitro. Furthermore, Pluronic® P123-coated PTX/LPB loaded Lps were evaluated for their cytotoxicity in MDA-MB 231 and MDA-MB 231/PTXR cells. In resistant MDA-MB 231/PTX cells, MDR-1-mediated resistance was examined using successfully coated Lps.
MATERIALS AND METHODS
Materials
The drug PTX was provided by MSN laboratories, Hyderabad, India, and the drug LPB was provided by Hetero drugs Pvt Ltd, Hyderabad, India. Sigma-Aldrich Co. (St Louis, MO) provided Pluronic® P123 (PEG20-PPO70-PEO20, molecular weight 5,800). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), maleimide-head group lipid 1,2-dio-leoyl-sn-glycero-3-phosphoeth-anolamine-N-[4-(p-maleimidophenyl) butyramide (MPB-PE), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DOPG), and cholesterol procured from Merck chemicals, Mumbai, India. We used fetal bovine serum (FBS), Dulbecco’s Modified Eagle Medium (DMEM), and other culture medium supplements supplied by Invitrogen Life Technologies (Carlsbad, CA); Falcon (Becton Dickinson, Franklin Lakes, New Jersey) supplied the plastic ware for cell cultures. Bio-Rad Laboratories (Hercules, CA) provided the electrophoresis reagents; Sigma Chemical Co. (St. Louis, MO) provided the Bicinchoninic acid kit for determining the protein content of cell monolayers and lysates. All other reagents were purchased from Sigma-Aldrich Co. unless otherwise specified.
Preparation of PTX and LPB dual-loaded Pluronic® P123-coated Lps
PTX and LPB dual-loaded Lps coated with Pluronic® P123 (PTX/LPB-PLps) are prepared by the conventional dehydration-rehydration method (Stolnik et al., 1994). Initially, DOPC, DOPG, and maleimide-head group lipid namely, MPB-PE were dissolved in chloroform at a molar ratio 1:0.25:1.25 (total lipid mixture) and added PTX and LPB at a molar ratio 1:0.33 to the lipid mixture. Using rotary vacuum evaporation, the organic solvent was removed from the phospholipid film and the final concentration of phospholipid reached 3.83 mg/ml by hydrating it in 300 mM ammonium-sulfate solution. The Lps experienced sonication in a bath at 50°C (200 W for 15 minutes) followed by probe sonication at 30 W for 5 minutes. (Sonics & Materials Inc., Danbury, CT). Dialyzing the external buffer with 0.9% NaCl solution (MW cut-off: 6,000–8,000) replaced the external buffer. During dialysis, fresh dialysate was replaced every 2 hours for a duration of 6 hours. The unencapsulated PTX and LPB were initially removed from the samples by centrifugation at 13,500 rpm for 5 minutes after incubation. The second step involved removing the supernatant and incubating Pluronic® P123 solution with Lps at a 1:1 mass ratio (Pluronic® P123: total lipid mixture with encapsulated drugs). A sephadex G-50 column (MicrofineTM analytical grade, GE Healthcare, UK) pre-equilibrated with phosphate-buffered saline (PBS) was used to eliminate leftover PTX and LPB and unabsorbed polymer from the mixture. In the control groups, empty Lps were compared with empty Lps coated with coated PLps, empty Lps loaded with PTX (PTX-Lps), LPB-Lps, and PTX-loaded Lps coated with Pluronic® P123 (PTX-PLps) and LPB-loaded Lps coated with Pluronic® P123 (PTX-PLps) and PTX-PLps were prepared in a similar manner. With the exception of the coating step, the same procedure was applied to the preparation of PTX/LPB-loaded Lps.
Characterization of Lps
A Malvern Nano Zetasizer ZS90 (Malvern Instruments, Worcestershire, UK) was used to determine liposome diameter, zeta potential, and polydispersity index (PDI). The characteristics of the liposome suspension were determined by diluting 10 µl of the suspension in 1 ml of 100 mM NaCl solution and analyzing it. The measurements were carried out at a temperature of 25°C at a fixed angle of 90° and employing a laser power of 40 mW. In order to measure the thickness of Pluronic® P123 coating, the uncoated particle size was subtracted from the coated particle size and the result was divided into two (Jiang et al., 2016). In order to visualize the morphology of the Lps, they were diluted at a concentration of 3.83 mg/ml in 0.9% w/v NaCl solution and analyzed with a transmission electron microscope (TEM, Model JEM-2100F, Tokyo, Japan) operating at an acceleration voltage of 80 k. Through high-performance liquid chromatography (HPLC), encapsulation efficiency and drug loading were determined. Acetonitrile was used as the solvent for dissolving the prepared Lps (liposome to ACN ratio = 1:9). A HPLC analytical system comprised of a 1,260 binary pump and an UV detector (Agilent Technologies, Santa Clara, CA) and a Zorbax® HPLC C18 column (250 × 4.6 mm; 5 µm) was used to quantify PTX. ACN: H2O (v/v) at 70:30 is used as the mobile phase (both ACN and H2O containing 0.1% trifluoroacetic acid). The analysis was initiated with an injection volume of 20 µl and analyzed at a flow rate of 1.0 ml/minutes, and at a column temperature of 25°C, and a UV detection wavelength of 227 nm. LPB was found to be in the same conditions as PTX, with the exception of a mobile phase composed of ACN: 10 mM ammonium acetate buffer: (80:20 v/v) with a UV detection wavelength of 272 nm (Saadat et al., 2016). The efficiency of PTX and LPB loading and encapsulation was estimated as follows.
Stability of Lps
We measured the diameter of particle size both in the presence of PBS buffer and 50% FBS as well as viscosity in order to demonstrate the stability of uncoated and coated PTX/LPB-loaded Lps (Wolfram et al., 2014). Formulations PTX/LPB-Lps and PTX/LPB-PLps were stored separately at 4°C in a refrigerator to measure the stability during storage. The 100 µl sample was diluted with PBS buffer (pH 7.4) to 1 ml at predetermined time points (0, 1, 2, 4, 6, 8, and 10 days) for diameter measurement using a Malvern Nano Zetasizer ZS90. We examined the stability of uncoated and coated PTX/LPB-Lps in hydrodynamic and serum conditions by mixing 3.83 µg/ml formulation with equal amounts of FBS and incubating them at 37°C for 30 minutes. By diluting 100 µl of the sample to 1 ml with PBS buffer (pH 7.4) at predetermined times (0, 2, 5, 9, 15, 21, and 30 hours), the samples were analyzed and the diameter was measured. Viscosity was determined on 10 ml of the prepared LPs using a Haake Rheo Stress 150 Rheometer type rotational viscometer. The measurement was carried out at 37°C at various angular velocities. During a typical run, the angular velocity was changed from 1 to 100 rpm at a controlled ramp speed. Viscosity values were recorded for every 20 seconds (Budai et al., 2007).
In-vitro drug release studies
Under sink conditions, a modified dialysis method was used to measure the in-vitro release of PTX, and LPB from Pluronic® P123 coated and uncoated single and dual drug-loaded Lps (Godwin et al., 1992). In one dialysis tube, 1 ml of Lps and 1 ml of FBS were introduced (Mw cut-off: 6,000–8,000), the two ends of which were clipped tightly together. A 50 ml EP tube containing 0.1% (w/v) Tween 80 was immersed in PBS (pH 7.4) containing the dialysis tube (Yang et al., 2007). The sample was then kept at 37°C for 48 hours while being shaken at 100 rpm (Lopez et al., 2011). At each of the specified intervals, 1 ml of the drug release medium was removed and substituted with an equal amount of fresh media (0.5, 1, 3, 5, 10, 24, 36, and 48 hours). The content of PTX and LPB was determined by HPLC analysis as described above. The same procedure is repeated to determine the amount of PTX, and LPB released from the physical mixture prepared at the established concentrations.
Cell culture
Human triple-negative breast cancer MDA-MB 231 and MDA-MB 231/PTX cells were cultured in a 5% CO2 environment with DMEM (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% FBS (Invitrogen Life Technologies (Carlsbad, CA) and 2 mM of L-glutamine (Invitrogen Life Technologies, Carlsbad, CA) as per the reported literature (Hero et al., 2019). In order to produce PTX-resistant MDA-MB 231 (MDA MB-231/PTX cells), MDA-MB 231 cells were continuously exposed to PTX at 40% inhibitory concentration (IC40 of 24 hours) for 4 days. The drug was then removed before the treatment was resumed. Following the interval of 4 weeks, the treatment was increased to the Half-maximal inhibitory concentration (IC50) level. The process was repeated until the cells were able to grow successfully under IC50 (24 hours) treatment of PTX. In the absence of PTX, the cells were no longer grown after they had been obtained. A fresh medium without PTX was then substituted for the old medium for a period of 7 days in order to recover the cells. Through the process of limiting dilution, where cells were serially diluted over 96 wells and then expanded, single cell-derived clones were obtained (Sun et al., 2011). These cells were then considered PTX-resistant MDA-MB 231 (MDA-MB 231/PTX cells).
In-vitro quantitative cellular uptake of PTX/LPB-PLps
In-vitro quantitative cellular accumulation of PTX/LPB was determined as per our earlier work published (Pitchika and Sahoo, 2022). For the purpose of quantifying the intracellular drug content, MDA-MB 231 and MDA-MB 231/PTX cells were seeded in 6-well plates at a density of 2 × 105 cells per well. A fresh medium was added to the medium after overnight incubation, and formulations, PTX-Lps, LPB-Lps, PTX-PLps, LPB-PLps, PTX/LPB-Lps, and PTX/LPB-PLps were added to achieve an ultimate concentration of PTX of 15 and 5 µg/ml, then incubated at 37°C for 3 hours. In order to remove unbound LPS from the cells, initially, growth media was withdrawn from the cells, followed by washing of cells twice with PBS. After centrifugation at 1,000 rpm to recover the cells, they were resuspended in PBS mixed with 1× triton-100 solution (Mittal et al., 2007). Using an ultrasonic lyser, the cells were lysed in a water bath for 5 minutes. In the same manner, as described in the preceding sections, HPLC analysis was used to determine the amount of PTX and LPB from selected preparations.
Observation of cellular morphology
One of the defining characteristics of tumor cells is abnormal cellular morphology. Cell morphology examines the shape, structure, form, and size of cells over the course of treatment. We performed the cell morphological observation over the treatment of selected Lps, as per the reported protocol (Bisht et al., 2016). After being cultivated (1 × 106 cells) in a 6-well plate, MDA-MB 231 and MDA-MB 231/PTX cells were treated for 24 hours with PTX/LPB dual loaded Pluronic® P123 coated and uncoated Lps, and their corresponding blank Lps. An Axio-Cam HRc CCD camera (Oberkochen, Germany) was used to acquire images of the cellular surface morphology of MDA-MB 231 and MDA-MB 231/PTX over the treatment.
In-vitro cytotoxicity
Using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, we were able to determine whether Lps and PLps formulations demonstrated cytotoxic activity against MDA-MB 231 and MDA-MB 231/PTX cells after post-incubation (24 and 48 hours) as per the method described earlier (Kunii et al., 2007). Briefly, the cells were placed in 96-well culture plates at a density of 4,000 to 6,500 cells per well. A fresh medium was added after overnight incubation, and all formulations (PTX-LPS, LPB-LPS, PTX-PLPs, LPB-PLPs, PTX/LPB-Lps, and PTX/LPB-PLps) were added to attain a total concentration of 90 nM for PTX and 30 nM for LPB. In order to determine the viability of the cells, all groups were exposed to the MTT reagent after 24 and 48 hours of incubation. Followed by the incubation, PTX-resistant MDA-MB 231/PTX cells and sensitive MDA-MB 231 cells were incubated for 4 hours in the presence of MTT (5 mg/ml). After the culture medium was discarded from the plates, dimethyl sulfoxide was used to dissolve the formazan crystals. Quantitative analysis was performed (Marslin et al., 2009) using a Multiwell Microtitre Spectrophotometer (Thermo Scientific MultiskanTM Microplate Photometer, Massachusetts, USA). Using the absorbance intensities of a control group, % viable cells were determined. A total of three replications were conducted for each experiment.
Western blot analysis
Expressional analysis of cellular key proteins, MDR1, kinase growth regulator EGFR, and its phosphorylated partners, was conducted (Mittal et al., 2007; Pasquier et al., 2013) in MDA-MB 231 and MDA-MB 231/PTX cell lines in order to elucidate the mechanisms for PTX and LPB release from PTX/LPB-PLps. A 6-well plate was seeded with 2 × 105 MDA-MB 231 and MDA-MB 231/PTX cells and permitted to attach to the culture plate for 6 hours. The cells were exposed to PTX/LPB-Lps and PTX/LPB-PLps for 12 hours after 24 hours of culture (at equivalent concentrations of PTX and LPB). In order to lyse the cells after treatment, they were washed in ice-cold lysis buffer containing 10% Triton-X100, 50 mM Tris, 10 mM ethylenediamine tetraacetic acid, and then cells were added with a protease inhibitor cocktail set III, 2 mM PMSF, and 1 mM Na3VO4 following sonication and centrifugation at 13,000 g. In addition to SDS-PAGE, approximately 25 µg of protein isolates were probed with antibodies of anti-MDR1/P-gp1, anti-EGFR, and anti-(phosphoTyr1248) EGFR from Invitrogen, Thermo Fisher Scientific, USA, and secondary antibodies with peroxidase conjugation (Cell Signaling Technologies, USA). Proteins were identified by enhanced chemiluminescence after the membranes had been rinsed with 0.1% Tris-buffered saline-Tween solution. The samples were probed with anti-tubulin antibodies (Cell Signaling Technologies, USA) in order to confirm that standards were loaded uniformly in lysates.
P-gp1 activity
The MDR activity of P-gp1 (MDR1) was determined using calcein-AM, a fluorescent anionic dye, as directed by the manufacturer’s protocol (Calcein-AM, Cayman Chemical, USA) as reported earlier (Pasquier et al., 2013). As the cytosolic esterase converts calcein acetoxymethyl ester (calcein-AM) into hydrophilic, intensely green fluorescent calcein, calcein-AM is a hydrophobic non-fluorescent dye that distributes in cells (Kabanov et al., 2003). A flat-bottomed 96-well black-wall microplate was used to grow MDA-MB 231 and MDA-MB 231/PTX cells at a density of 1 × 104 cells per well for 24 hours in 100 µl of complete growth medium. After 12 hours of treatment with PTX/LPB-Lps and PTX/LPB-PLps (at equivalent concentrations of PTX/LPB, 45 and 15 nM, respectively), the cells were washed with PBS. The selected formulations were pre-treated with 100 µl of the calcein-AM solution and then incubated at 37°C for 30 minutes in the dark. In order to measure intracellular fluorescence, a microplate reader (Spark® reader, Tecan, USA) set to excitation λmax of 485 nm and emission λmax of 535 nm was used. In comparison with control wells, the functional activity of P-gp1 was determined as a % of fluorescence detected.
Statistical analysis
In the text and Figs, all data are presented as Mean ± SD. ANOVA and Tukey’s test were used to analyze the results using Graph-Pad Prism (version 6.01). It was considered significant if the p-value was less than 0.05.
RESULTS AND DISCUSSION
Characterization of Lps
Initially, we designed and successfully prepared PTX alone and LPB alone loaded Lps, as well as PTX/LPB dual loaded Lps with and without Pluronic® P123 coating. Following thin film hydration and ammonium sulfate gradient preparation, each formulation was prepared either using the coating or without coating. According to results shown in Table 1, single drug-loaded Lps, such as those containing PTX (PTX-Lps) and LPB (LPB-Lps), were measured to have particle sizes of 123.54 ± 4.35 and 121.97 ± 5.33 nm respectively and their PDI was in the range of 0.26 to 0.29. PTX/LPB dual-loaded Lps showed an increased particle size of 165.35 ± 4.82 nm and a stable PDI of 0.28. As predicted, the average particle size increased after incubation with Pluronic® P123 (136.69 ± 3.91, 139.42 ± 6.17, and 183.41 ± 4.96 nm for PTX-PLps, LPB-PLps, and PTX/LPB-PLps, respectively). The mechanism of Pluronic® P123 adsorption onto the liposomal surface involves the hydrophobic interactions (non-covalent) between the polymeric surfactant and the lipids that comprise the liposome membrane. When pluronic polymer is added to an aqueous solution containing Lps, the hydrophobic PPO blocks of Pluronic are attracted to the hydrophobic regions of the liposome membrane, while the hydrophilic PEO blocks extend into the aqueous medium, forming a stable and protective coating around the Lps. The adsorption of Pluronic onto the liposomal surface is driven by the difference in free energy between the Pluronic-lipid and Pluronic-water interfaces. The hydrophobic interactions between the PPO blocks of Pluronic and the lipid molecules of the liposome result in a decrease in free energy, which promotes the adsorption process (Batrakova et al., 2001; Vu-Quang et al., 2016). The coating layers measured between 11.24 and 17.69 nm for PTX-PLps, LPB-PLps, and PTX/LPB-PLps, which is higher than that documented earlier for poloxamer absorbed to PLGA nanoparticles (3–6 nm) (Batrakova et al., 2001). The formation of a Pluronic® P123 coating layer can be seen in the fact that the PDI value of coated LPB/PTX-Lps (PTX/LPB-PLps) was higher (0.32 to 0.35) than the value of uncoated Lps. The surface charge of uncoated and coated Lps was similarly negative (2.7 to 3.6 mV) when considered from the perspective of Zeta potential. % encapsulation (% EE) and % drug loading (% DL) of PTX and LPB of coated Lps were slightly higher than that observed for uncoated Lps (at % higher EE of 4.15, and 5.06, for PTX and LPB, respectively, in single drug loaded Lps and at % higher EE of 4.54, and 4.29 for PTX and LPB respectively in dual loaded Lps; % higher DL was reported in the range of 0.51 to 1.06 among coated and uncoated Lps as shown in Table 2). During liposome dialysis, a small amount of PTX and LPB, which was on or near the exterior of uncoated Lps, was quickly released. The results also demonstrated that dual-loaded Lps and PLps reported slightly lower % EE and % DL compared with their single-drug-loaded counterparts.
The morphology of single and dual drug-loaded Pluronic® P123 coated, and uncoated Lps (PTX-Lps, LPB-Lps, PTX-PLps, LPB-PLps, PTX/LPB-Lps, and PTX/LPB-PLps) were analyzed by TEM. Images of both coated and uncoated Lps indicated that the former had smooth spherical forms, whereas the latter displayed relatively rough surfaces after being coated with Pluronic® P123 (Fig. 1). The TEM analysis also confirmed that Pluronic® P123 coated Lps absorb the coating on their exterior by confirming the increased particle size of PLps. The smooth surface of Lps has been shown to be less irritating to tissue than those with a crystalline or irregular surface (Hu et al., 2015). DLS was used to evaluate the storage stability of Pluronic® P123 uncoated and coated PTX/LPB dual-loaded Lps in aqueous and serum conditions. As demonstrated in Figure 2A and B, the hydrodynamic dimensions of PTX/LPB-Lps and PTX/LPB-PLps did not change significantly after aqueous storage at 4°C (over 10 days) or in 50% serum conditions (over 0 to 30 days). These results were supported by the particle size distribution of PTX/LPB-Lps and PTX/LPB-PLps measured by DLS during the end of the storage period (Fig. 2C and D). PTX/LPB-Lps and PTX/LPB-PLps displayed consistent particle size ranges from 160 to 189 nm. These results support the stable determination of size distribution. The viscosity of liposomal formulations is primarily determined by the size, concentration, and composition of the lipids that are used to form Lps. Due to the additional polymer chains in the formulation, Pluronic P123 coating slightly increased the viscosity of the LPs, however, the effect of coating is negligible when compared to the uncoated LPs formulation (Fig. 2e). Coated Lps are also characterized by their excellent stability because of the hydrophilic PEO chain of Pluronic® P123 that is exposed on their exterior. Figure 3 showed the in-vitro release profiles of uncoated single drug-loaded PTX and LPB Lps (PTX-Lps, and LPB-Lps), uncoated dual-loaded PTX/LPB-Lps and Pluronic® P123 coated single drug loaded PTX, and LPB Lps (PTX-PLps, and LPB-PLps) coated dual loaded PTX/LPB-PLps as well as a physical mixture containing equivalent amounts of PTX and LPB. Results revealed that the % in-vitro release of LPB was superior to PTX regardless of coating or single or dual drug loading process. As shown in Figure 3a, Pluronic® P123 coated Lps (PTX-PLps and LPB-PLps) exhibited a higher % release of both PTX and LPB when compared to uncoated PTX-Lps and LPB-Lps. As determined by in-vitro release profiles, both PTX and LPB were continuously released from Lps without any primary burst release effect, and the cumulative amount of PTX and LPB released from single drug-loaded PLps over 48 hours were approximately 82.91% ± 5.23% and 99.34% ± 5.81% respectively. Both PTX and LPB from the physical mixture demonstrated poor in-vitro release over a 48 hours period, i.e. 33.96 ± 2.17, and 55.13 ± 3.04, respectively. Further results revealed that both uncoated and coated PTX/LPB dual-loaded Lps showed a continuous rise in the release pattern of both PTX and LPB from Lps without any initial burst release effect (Fig. 3b and c). Interestingly, the codelivery of LPB has significantly increased the in-vitro release of PTX from dual-loaded PTX/LPB Lps (15.22%) as well as PTX/LPB-PLps (14.07%) over 48 as shown in Figure 3b and c. Results also demonstrated that dual-loaded PTX/LPB Lps with Pluronic® P123 coating has increased the in-vitro release of both PTX and LPB substantially by 14.07% and 8.48%, respectively. In Figure 3b and c, the PTX and LPB from PTX/LPB-PLps showed a maximum release of 93.69% and 99.95%, respectively, over the course of 48 hours.
 | Table 1. Particle size, zeta potential, and PDI data for Lps. [Click here to view] |
 | Table 2. % Encapsulation and % loading of PTX and LPB into Lps. [Click here to view] |
 | Figure 1. TEM images of Pluronic® P123 uncoated and coated PTX, and LPB single and dual loaded Lps (From right to left of upper row PTX-Lps, LPB-Lps, PTX/LPB-Lps and from right to left of lower row PTX-PLps, LPB-PLps, and PTX/LPB-PLps); Bar = 50 nm. [Click here to view] |
In vitro quantitative cellular uptake of Lps
Figure 4 displayed Pluronic® P123-coated and uncoated single and dual drug-loaded Lps were evaluated for cellular uptake of PTX and LPB following a 4-hour incubation post-treatment in MDA-MB 231 and MDA-MB 231/PTX cells. The cellular uptake and absorption of PTX and LPB from Pluronic® P123 coated single and dual-loaded Lps were significantly increased in MDA-MB 231 and MDA-MB 231/PTX cells compared to uncoated Lps. As a consequence of prolonged interaction between surface-coated Pluronic® P123 Lps and the cell membrane, the highest release of drug-carrier complexes into the cytosol was observed. The cellular uptake of PTX from Pluronic® P123-coated and uncoated Lps differed significantly between MDA-MB 231 and MDA-MB 231/PTX cells. PTX and LPB accumulation from Pluronic® P123-coated PTX/LPB-PLps in MDA-MB 231/PTX cells were 52.17% ± 2.8% and 73.64% ± 3.2%, respectively, which were different from PTX (55.74% ± 4.2%) and LPB (74.82% ± 3.8%) accumulation, reported in MDA-MB 231 cells. Furthermore, it was demonstrated that LPB accumulation was comparable regardless of the cell type (weather-sensitive or resistant MDA-MB 231 cells) or whether the Lps were loaded with a single or dual drug. But the pluronic® P123 coating significantly affected the % accumulation of PTX and LPB in both MDA-MB 231 and MDA-MB 231/PTX cells. % PTX and LPB accumulation due to pluronic® P123 coating was increased to 12.16% and 23.98% respectively in MDA-MB 231 cells, and about 22.53% and 22.42% increased accumulation of PTX, and LPB was observed in MDA-MB 231/PTX cells. Following 4 hours of post-treatment incubation, PTX and LPB delivered by dual-loaded PLps were considerably greater (p < 0.05) than those delivered by Pluronic® P123 uncoated Lps. As a consequence of these findings, it is evident from the in vitro drug release studies that co-delivery of LPB significantly increases the accumulation of PTX in both Pluronic® P123 coated and uncoated Lps. According to these results, Pluronic® P123 coating significantly accelerated the accumulation of both PTX and LPB from a variety of formulations regardless of the type of cell type (sensitive or resistant MDA-MB 231 cells) or whether the LP was single or dual-loaded.
 | Figure 2. Stability studies of PTX/LPB-Lps and PTX/LPB-PLps; (a) Particle size changes of PTX/LPB-Lps and PTX/LPB-PLps in PBS buffer. (b) Particle size changes of PTX/LPB-Lps and PTX/LPB-PLps in 50% FBS solution. Particle size distribution of PTX/LPB-Lps (c) and PTX/LPB-PLps (d); and the viscosity of prepared LPs with or without pluronic coating (e) during the storage period. Results were represented as mean ± SD (n = 3). [Click here to view] |
Cellular morphologic changes over treatment of Lps
PTX/LPB-loaded Pluronic® P123 coated Lps treatment significantly changed the morphological behavior of both MDA-MB 231 and MDA-MB 231/PTX cells at 24 hours when compared with uncoated PTX/LPB Lps. As a consequence of being incubated with dual-loaded Pluronic® P123 coated Lps containing 9 and 3 µg/ml of PTX and LPB, respectively, long spindle-shaped cells rounded off, and intercellular spaces enlarged. Furthermore, the refraction of the cells in the treated groups increased (Fig. 5). The effect of cellular rounding was clearly evident in coated Lps as compared to uncoated Lps. Therefore, these results demonstrate that Pluronic® P123 coated Lps cause morphological deformation of MDA-MB 231 and MDA-MB 231/PTX cells at their established concentrations.
In-vitro cytotoxicity studies
PTX and LPB Lps with or without Pluronic® P123 coating, along with the blank Lps, were assessed for their cytotoxicity against MDA-MB 231 and MDA-MB 231/PTX cells. In order to test the therapeutic efficacy of various treatments, the cells were initially divided into eight groups and incubated for 24 and 48 hours. Different formulations of single or dual drug-loaded LPS containing 90 and 30 nM of either PTX or LPB, or both, were tested. Figure 6 illustrates the almost negligible cytotoxicity of blank (Lps coated with Pluronic® P123 coating) in both MDA-MB 231 and MDA-MB 231/PTX cells. Uncoated PTX and LPB alone Lps suppressed MDA-MB 231 cell proliferation by 36.43% and 37.89%, respectively, after 24 and 48 hours of incubation. Uncoated PTX alone, Lps exerted a poor cytotoxic response in MDA-MB 231/PTX cells, with cytotoxicity of 12.61% and 13.44% at 24 and 48 hours, respectively. However, data showed that uncoated LPB-Lps had a substantially equal impact on PTX-sensitive and -resistant MDA-MB 231 cells. Based on the results of this investigation, it was also assessed that MDA-MB 231/PTX cells were resistant to PTX-Lps but susceptible to LPB-Lps, and liposomal delivery did not significantly affect the cytotoxic effectiveness of PTX alone treatment. PTX-loaded Lps coated with Pluronic® P123 showed significantly increased cytotoxic effects against MDA-MB 231 and MDA-MB 231/PTX cells by 8.3% and 12.82%, respectively, at 24 hours. It was found that LPB-loaded Lps coated with Pluronic® P123 exhibited a slight increase in cytotoxicity both in sensitive and resistant MDA-MB 231 cells. PTX and LPB co-delivery induced a greater cell-killing effect, as demonstrated by results in both MDA-MB 231 and MDA-MB 231/PTX cells as compared with the corresponding PTX or LPB alone. By encapsulating dual agents in a liposomal system, the potency of the formulation was significantly increased, as indicated by in-vitro drug accumulation studies. Uncoated PTX/LPB dual-loaded Lps exhibited a significant cytotoxic effect of 68.38% ± 4.14% and 64.26% ± 4.36% in MDA-MB 231 and MDA-MB 231/PTX cells, respectively, after 24 hours of treatment. PTX/LPB dual-loaded Lps coated with Pluronic® P123 demonstrated substantial cytotoxicity in MDA-MB 231 and MDA-MB 231/PTX cells after 24 hours of treatment, with a rate of 88.57% ± 5.27% and 85.33% ± 5.11%, respectively (Fig. 6). These results demonstrated that the Pluronic® P123 coating significantly impacted the delivery of two drugs to the cancer microenvironment. Therefore, Pluronic® P123-coated liposomal encapsulation of PTX and LPB could increase drug-induced cytotoxicity against both MDA-MB 231 and MDA-MB-231/PTX cells, to a considerable extent, than uncoated Lps.
 | Figure 3. In-vitro release profiles of PTX and LPB from PTX-Lps, LPB-Lps, PTX-PLps, LPB-PLps and Physical mixture (a); and from uncoated dual loaded PTX/LPB-Lps (b) and P123 coated PTX/LPB dual loaded Lps in PBS (pH 7.4) with 0.1% (v/v) Tween 80. Results (n = 3) were shown as a mean ± SD. [Click here to view] |
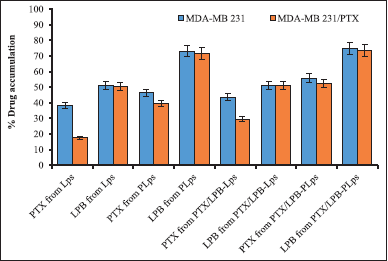 | Figure 4. A quantitative study of the cellular uptake of PTX/LPB-loaded Pluronic® P123 coated Lps in MDA-MB 231 and MDA-MB 231/PTX cells was conducted. The results are expressed as mean ± SD (n = 3). p < 0.05 (*) indicated statistical significance compared with uncoated Lps. [Click here to view] |
 | Figure 5. Morphologic changes associated with MDA-MB 231 and MDA-MB 231/PTX cells over dual PTX/LPB loaded Pluronic® P123 coated Lps and dual PTX/LPB loaded uncoated Lps at 24 hours of incubation. (a) MDA-MB 231 control cells (b) MDA-MB 231/PTX control cells; (c) Treatment of uncoated PTX/LPB loaded Lps in MDA-MB 231 and (d) MDA-MB 231/PTX cells; (e) Treatment of coated PTX/LPB loaded Lps with equivalent quantity in MDA-MB 231 and (f) MDA-MB 231/PTX cells. [Click here to view] |
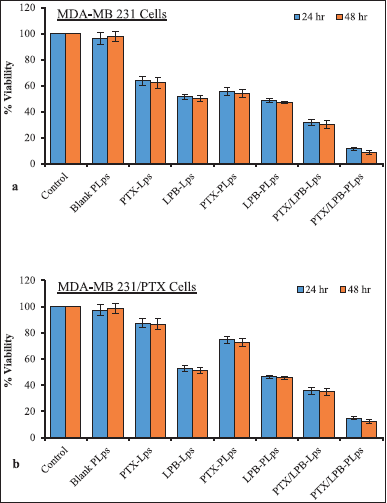 | Figure 6. MTT assay was carried out to determine the potential cytotoxic efficiencies of the treatments on MDA-MB 231 cells (a) and MDA-MB 231/PTX cells (b); Data were presented as mean ± SD (n = 3). [Click here to view] |
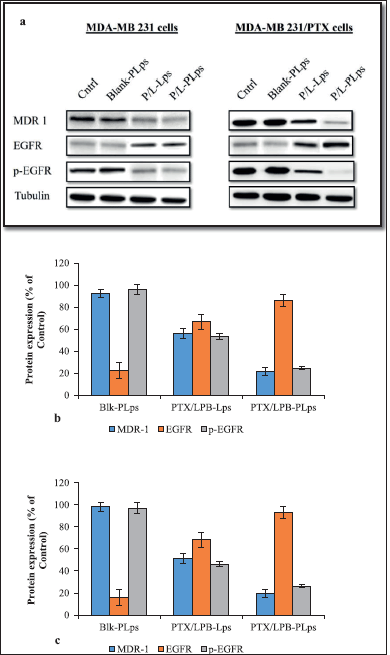 | Figure 7. As a result of incubation with PTX/LPB dual-loaded LPS coated and uncoated, as well as blank LPS, MDA-MB 231 and MDA-MB 231/PTX cells expressed various levels of MDR-1, EGFR, and p-EGFR; Western blot analysis demonstrating the expression profiles of protein targets in MDA-MB 231 and MDA-MB 231/PTX cells after treatment (a); quantitative measurement of the target protein levels in MDA-MB 231 cells when compared with the control (b); in MDA-MB 231/PTX cells when compared with the control (c); The quantitative data were expressed as a percentage that was determined by analyzing how to target protein expression was expressed in control cells (n = 3, mean ± SD). [Click here to view] |
PTX/LPB loaded-Pluronic® P123 coated Lps downregulate MDR1-mediated EGFR signaling
This study investigated the production of endogenous MDR1, EGFR, and phosphorylated EGFR (p-EGFR) in MDA-MB 231 and MDA-MB 231/PTX cells after being treated with Pluronic® P123 coated or uncoated PTX/LPB loaded Lps to further understand MDR-1 mediated EGFR mechanism. Antibodies targeting MDR1, EGFR, and p-EGFR were incubated with protein lysate and membranes after treatment. In contrast to uncoated PTX/LPB dual-loaded Lps, Pluronic® P123 coated treatment dramatically decreased the MDR1 and phosphorylated EGFR levels in MDA-MB 231 as well as in MDA-MB 231/PTX cells, as shown by their protein expression data in Figure 7. The results also revealed that the coated Lps had a greater effect on MDA-MB 231/PTX cells than on MDA-MB 231 cells. The expression of EGFR was increased in Pluronic® P123 coated and uncoated PTX/LPB-loaded Lps, which confirms that blocking of EGFR phosphorylation is accomplished by the treatment It was found that coated Lps resulted in the greatest improvement in EGFR expression in both MDA-MB 231 and MDA231/PTX cells when compared to uncoated Lps. As in the aforementioned results, blank Lps and PLps had similar effects as MDA-MB 231 and MDA-MB 231/PTX control cells, indicating no change in the levels of MDR1, and EGFR target proteins (Fig. 7). Based on the data presented here, coated Lps significantly inhibited MDR1-mediated EGFR signaling in MDA-MB 231 and MDA-MB 231/PTX cells (Fig. 7b and c showed a p < 0.05 value for coated Lps in comparison to untreated control cells).
Inhibition of P-gp1 activity
Pluronic® P123 coated Lps loaded with PTX/LPB to combat MDR1-driven PTX resistance in MDA-MB 231/PTX cells were evaluated using the calcein-AM assay. The kit contains calcein-AM, a non-fluorescent P-gp1 substrate that, upon cytoplasmic cleavage, becomes the fluorescent molecule calcein. Figure 8 displayed the outcomes of the P-gp1 activity assessments. As a first step toward comprehending MDR1-associated PTX resistance in MDA-MB 231/PTX cells, P-gp1 activity was analyzed by comparing the fluorescence intensity of calcein in MDA-MB 231 and MDA-MB 231/PTX cells. The results showed that MDA-MB 231/PTX cells, which have a reduced capacity to ingest calcein due to P-gp1 activity in the cytoplasm, had the lowest intensity compared to MDA-MB 231 cells. A similar lack of response was observed when uncoated PTX/LPB loaded Lps were treated, resulting in elevated intracellular calcein fluorescence (Fig. 8). Coated PTX/LPB Lps significantly lowered P-gp1 activity with a rise in fluorescence intensity of 90%, whereas the combination of PTX and LPB in either uncoated or coated liposomal form boosted intracellular calcein fluorescence in both MDA-MB 231 and MDA-MB 231/PTX cells. Using MDR1 suppression, this work showed that a combination of PTX and LPB, especially when placed into a Pluronic® P123 coating, can reduce the lifespan of triple-negative MDA-MB 231 and PTX-resistant triple-negative breast cancer cells.
 | Figure 8. Effect of coated and uncoated PTX/LPB Lps over P-gp1 activity in MDA-MB 231 and MDA-MB 231/PTX breast cancer cells. Cells were treated with coated and uncoated PTX/LPB Lps for 1 hour, and then calcein-AM assay was performed. Calcein fluorescence was expressed as a percentage of the control (mean ± SD, n = 4). * p < 0.05 versus. PTX/LPB binary mixture. [Click here to view] |
CONCLUSION
A new Pluronic® P123 coated PTX and LPB liposomal formulation were devised for the first time in this study for its ability to decrease P-gp1 expression and rescue PTX resistance, as well as improve its efficacy against resistant triple negative breast cancer cells. The Pluronic® P123-coated and uncoated PTX and LPB dual-loaded Lps were prepared by loading PTX and LPB at the desired mass ratios and were successfully characterized. Dual drug-loaded coated Lps showed improved in vitro release of PTX, and LPB demonstrated rapid release of PTX remarkably in combination with LPB in an environment similar to that found within tumor cells. Moreover, compared with uncoated dual-loaded Lps, their combination with Pluronic® P123 coating enhanced cytotoxicity synergistically by inhibiting MDR1-mediated resistance in MDA-MB 231 and MDA-MB 231/PTX cells. Due to their limited selectivity, potential toxicity, and suppression of P-gp1 in non-transformed tissues, all P-gp1 inhibitors have consistently failed, rendering these findings, particularly significant (Engle and Kumar, 2022). Despite the fact that triple-negative breast cancer expresses large levels of P-gp1, it is resistant to a wide range of chemotherapeutic treatments. There is still a need for more effective chemotherapy for treating breast cancers of the triple-negative subtype. The clinical relevance of our findings stems from the fact that adjuvant chemotherapy is one of the most cutting-edge treatment choices for triple-negative breast cancer. Our research suggests that Lps that is PTX/LPB dual-loaded with Pluronic® P123 coating might aid in this effort. Patients with breast cancer that has become resistant to many drugs may benefit from a clinical trial using the combination of PTX/ LPB Lps.
ACKNOWLEDGMENT
The authors acknowledge the Research Lab, School of Pharmacy, Gitam University, for providing the necessary facilities to carry out the research activities. The authors are thankful to Mr. Sanjay Kumar, research scholar, GITAM School of Pharmacy, Gitam University, for his support, and Dr. G. Shiva Kumar, Professor, Gitam University, Hyderabad, for their valuable suggestions and analysis of the results.
AUTHOR CONTRIBUTIONS
Suvendu Kumar Sahoo has designed, and supervised the execution of work, and contributed to manuscript revision. Subrahmanyam Pitchika performed the experiments, interpreted the analysis data, and contributed to the manuscript writing.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ajani JA, Ilson DH, Daugherty K, Pazdur R, Lynch PM, Kelsen DP. Activity of taxol in patients with squamous cell carcinoma and adenocarcinoma of the esophagus. J Nat Cancer Inst, 1994; 86(14):1086–91.
Akbarzadeh A, Rezaei SA, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Koshki KN. Liposome: classification, preparation, and applications. Nanoscale Res Lett, 2013; 8(1):1254–63.
Anirudhan TS, Varghese S, Manjusha V. Hyaluronic acid coated pluronic F127/pluronic P123 mixed micelle for targeted delivery of paclitaxel and curcumin. Int J Biol Macromol, 2021; 192:950–7.
Baselga J, Albanell J, Ruiz A, Lluch A, Gascon P, Guillem V, González S, Sauleda S, Marimón I, Tabernero JM, Koehler MT, Rojo F. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J Clin Oncol, 2005; 23(23):5323–33.
Batrakova EV, Li S, Vinogradov SV, Alakhov VY, Miller DW, Kabanov AV. Mechanism of pluronic effect on P-glycoprotein efflux system in blood-brain barrier: contributions of energy depletion and membrane fluidization. J Pharmacol Exp Ther, 2001; 299(2):483–93.
Bisht S, Schlesinger M, Rupp A, Schubert R, Nolting J, Wenzel J, Holdenrieder S, Brossart P, Bendas G, Feldmann G. A liposomal formulation of the synthetic curcumin analog EF24 (Lipo-EF24) inhibits pancreatic cancer progression: towards future combination therapies. J Nanobiotechnol, 2016; 14:57.
Budai L, Hajdu M, Budai M, Grof P, Beni S, Noszal B, Klebovich I, Antal I. Gels and liposomes in optimized ocular drug delivery: studies on ciprofloxacin formulations. Int J Pharm, 2007; 343(1–2):34–40.
Cruz LJ, Tacken PJ, Bonetto F, Buschow SI, Croes HJ, Wijers M, De vries MJ, Figdor CJ. Multimodal imaging of nanovaccine carriers targeted to human dendritic cells. Mol Pharm, 2011; 8(2):520–31.
Cruz LZ, Tacken PZ, Eich C, Rueda F, Torensma R, Figdor CG. Controlled release of antigen and toll-like receptor ligands from PLGA nanoparticles enhances immunogenicity. Nanomedicine, 2017; 12(5):491–510.
Cruz LJ, Tacken PJ, Zeelenberg IS, Srinivas M, Bonetto F, Weigelin B, Eich C, de vries IJ, Figdor CG. Tracking targeted bimodal nanovaccines: immune responses and routing in cells, tissue, and whole organism. Mol Pharm, 2014; 11(12):4299–313.
Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J, Shi B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res, 2015; 5(10):2929–43.
Dickler MN, Cobleigh MA, Miller KD, Klein PM, Winer EP. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res Treat, 2009; 115(1):115–21.
Engebraaten O, Vollan HKM, Børresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol, 2013; 183(4):1064–74; doi: 10.1016/j.ajpath.2013.05.033.
Engle K, Kumar G. Cancer multidrug-resistance reversal by ABCB1 inhibition: a recent update. Eur J Med Chem, 2022; 239:114542.
Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat, 2016; 26:1–9.
Furstova VN, Kopperova D, Balusikova K, Ehrlichova M, Brynychova V, Vaclavikova R, Daniel P, Soucek P, Kovar J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol Appl Pharmacol, 2016; 310:215–28.
Gangi A, Chung A, Mirocha J, Liou DZ, Leong T, Giuliano AE. Breast-conserving therapy for triple-negative breast cancer. JAMA Surg, 2014; 149(3):252–8.
Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA, 1992; 89:3070–4.
Hero T, Bühler H, Kouam PN, Priesch GB, Lateit T, Adamietz IA. The triple-negative breast cancer cell line MDA-MB 231 is specifically inhibited by the ionophore salinomycin. Anticancer Res, 2019; 39(6):2821–7.
Hu H, Lin Z, He B, Dai W, Wang X, Wang J, Zhang X, Zhang H, Zhang Q. A novel localized co-delivery system with lapatinib microparticles and paclitaxel nanoparticles in a peritumorally injectable in situ hydrogel. J Control Release, 2015; 220(Pt A):189–200.
Illum L, Davis SS. The organ uptake of intravenously administered colloidal particles can be altered using a non-ionic surfactant (Poloxamer 338). FEBS Lett, 1984; 167(1):79–82.
Jiang L, He B, Pan D, Luo K, Yi Q, Gu Z. Anti-cancer efficacy of paclitaxel loaded in pH triggered liposomes. J Biomed Nanotechnol, 2016; 12(1):79–90.
Kabanov AV, Batrakova EV, Miller DW. Pluronic block copolymers as modulators of drug efflux transporter activity in the blood-brain barrier. Adv Drug Deliv Rev, 2003; 55(1):151–64.
Kato K, Tahara M, Hironaka S, Muro K, Takiuchi H, Hamamoto Y, Imamoto H, Amano N, Seriu T. A phase II study of PTX by weekly 1-h infusion for advanced or recurrent esophageal cancer in patients who had previously received platinum-based chemotherapy. Cancer Chemother Pharmacol, 2011; 67(6):1265–72.
Kavallaris M. Microtubules and resistance to tubulin binding agents. Nat Rev Cancer, 2010; 10(3):194–204.
Kunii R, Onishi H, Machida Y. Preparation and anti-tumor characteristics of PLA/(PEG-PPG-PEG) nanoparticles loaded with camptothecin. Eur J Pharm Biopharm, 2007; 67(1):9–17.
Levit SL, Yang H, Tang C. Rapid self-assembly of polymer nanoparticles for synergistic codelivery of paclitaxel and lapatinib via flash nanoprecipitation. Nanomaterials, 2020; 10(3):563–74.
Lin Y, He X, Zhou D, Li L, Sun J, Jiang X. Co-delivery of doxorubicin and itraconazole by Pluronic® P123 coated liposomes to enhance the anticancer effect in breast cancers. RSC Adv, 2018; 8(42):23768–79.
Liu Y, Ren Z, Yuan L, Xu S, Yao Z, Qiao L, Li K. Paclitaxel plus cisplatin vs. 5-fluorouracil plus cisplatin as first-line treatment for patients with advanced squamous cell esophageal cancer. Am J Cancer Res, 2016; 6(10):2345–50.
Lopez GP, Iglesias I, Benedi J, Lozano R, Teijon JM, Blanco MD. Paclitaxel-loaded polyester nanoparticles prepared by spray-drying technology: in vitro bioactivity evaluation. J Microencapsul, 2011; 28:417–29.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer, 2016; 16(2):110–20.
Marslin G, Sheeba CJ, Kalaichelvan VK, Manavalan R, Reddy PN, Franklin G. Poly(D,L-lactic-co-glycolic acid) nanoencapsulation reduces Erlotinib-induced subacute toxicity in rat. J Biomed Nanotechnol, 2009; 5(5):464–71.
Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther, 2008; 30(8):1426–47.
Mi FL, Wang LF, Chu PY, Peng SL, Feng CL, Lai YJ, Li JN, Lin YH. Active tumor-targeted co-delivery of epigallocatechin gallate and doxorubicin in nanoparticles for combination gastric cancer therapy. ACS Biomater Sci Eng, 2018; 4(8):2847–59.
Mittal G, Sahana DK, Bhardwaj V, Kumar MN. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Control Release, 2007; 119(1):77–85.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, Gilks CB, van de Rijn M, Perou CM. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res, 2004; 10(16):5367–74.
Pasquier J, Rioult D, Abu-Kaoud N, Marie S, Rafii A, Guerrouahen BS, Foll FL. P-glycoprotein-activity measurements in multidrug resistant cell lines: single-cell versus single-well population fluorescence methods. Biomed Res Int, 2013; 2013:676845.
Pitchika, S, Sahoo, SK. Paclitaxel and lapatinib dual loaded chitosan-coated PLGA nanoparticles enhance cytotoxicity by circumventing MDR1-mediated trastuzumab resistance in HER2 positive breast cancers: in-vitro and in-vivo studies. J Drug Deliv Sci Technol, 2022; 73:103445.
Redhead HM, Davis SS, Illum L. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation. J Control Release, 2001; 70(3):353–63.
Saadat E, Ravar F, Dehghankelishadi P, Dorkoosh FA. Development and validation of a rapid RP-HPLC-DAD analysis method for the simultaneous quantitation of paclitaxel and lapatinib in a polymeric micelle formulation. Sci Pharm, 2016; 84(2):333–45.
Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics, 2008; 9(1):105–27.
Stolnik S, Dunn SE, Garnett MC, Davies MC, Coombes AG, Taylor DC, Irving MP, Purkiss SC, Tadros TF, Davis SS, Illum L. Surface modification of poly(lactide-co-glycolide) nanospheres by biodegradable poly(lactide)-poly(ethylene glycol) copolymers. Pharm Res, 1994; 11(12):1800–8.
Sun G, Hsueh PY, Janib SM, Hamm-Alvarez S, MacKay JA. Design and cellular internalization of genetically engineered polypeptide nanoparticles displaying adenovirus knob domain. J Control Release, 2011; 155(2):218–26.
Vergara D, Bellomo C, Zhang X, Vergaro V, Tinelli A, Lorusso V, Rinaldi R, Lvov YM, Leporatti S, Maffia M. Lapatinib/Paclitaxel polyelectrolyte nanocapsules for overcoming multidrug resistance in ovarian cancer. Nanomedicine, 2012; 8(6):891–9.
Vu-Quang H, Vinding MS, Xia D, Nielsen T, Ullisch MG, Dong M, Nielsen NC, Kjems J. Chitosan-coated poly(lactic-co-glycolic acid) perfluorooctyl bromide nanoparticles for cell labeling in (19)F magnetic resonance imaging. Carbohydr Polym, 2016; 136:936–44.
Wind NS, Holen I. Multidrug resistance in breast cancer: from in vitro models to clinical studies. Int J Breast Cancer, 2011; 24:1–12.
Wolfram J, Suri K, Huang Y, Molinaro R, Borsoi C, Scott B, Boom K, Paolino D, Fresta M, Wang J, Ferrari M, Celia C, Shen H. Evaluation of anticancer activity of celastrol liposomes in prostate cancer cells. J Microencapsul, 2014; 31(5):501–7.
Xu L, Zhao Z, Wang K, Zhou H, Xing C. Expression of aldehyde dehydrogenase 1 and ATP-binding cassette superfamily G member 2 is enhanced in primary foci and metastatic lymph node from patients with triple-negative breast cancer. Biomed Res, 2017; 28:5078–83.
Yamada A, Ishikawa T, Ota I, Kimura M, Shimizu D, Tanabe M, Chishima T, Sasaki T, Ichikawa Y, Morita S, Yoshiura KI, Takabe K, Endo I. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res Treat, 2013; 137(3):773–82.
Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, Kim DD. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm, 2007; 338:317–26.
Zacharoula I, Athina A, Efstathia V, Konstantinos A, Constantinos T. Star-Graft quarterpolymer-based polymersomes as nanocarriers for co-delivery of hydrophilic/hydrophobic chemotherapeutic agents. ACS Omega, 2018; 3(9):11896–908.
https://www.cancer.org/cancer/breast-cancer/about/types-of-breast-cancer/triple-negative.html (Accessed 14 November 2012)
https://www.who.int/news-room/fact-sheets/detail/breast-cancer (Accessed 14 November 2012)